SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 20-F
☐ REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934
Or
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2016
Or
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
Or
☐ SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
Commission File No. 001-36203
Can-Fite BioPharma Ltd.
(Exact name of Registrant as specified in its charter)
Can-Fite BioPharma Ltd., an Israeli Limited Company
(Translation of the Registrant’s name into English)
Israel
(Jurisdiction of incorporation)
10 Bareket Street,
Kiryat Matalon,
P.O. Box 7537,
Petah-Tikva
4951778, Israel
(Address of principal executive offices)
Motti Farbstein
Chief Operating and Financial Officer
Tel: +972 (3) 924-1114
Fax: +972 (3) 924-9378
motti@canfite.co.il
10 Bareket Street,
Kiryat Matalon,
P.O. Box 7537,
Petah-Tikva
4951778, Israel
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
American Depositary Shares, each representing 2 Ordinary Shares, par value NIS 0.25 per share
(Title of Class)
Ordinary Shares, par value NIS 0.25 per share*
(Title of Class)
Securities registered or to be registered pursuant to Section 12(g) of the Act:
None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act:
None
* Not for trading, but only in connection with the registration of the American Depositary Shares.
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report (December 31, 2016): 27,709,901 are issued and outstanding (excluding 446,827 ordinary shares held as treasury shares).
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such a shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☐ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one): Large accelerated filer ☐ Accelerated filer ☐ Non-accelerated filer ☒
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| U.S. GAAP ☐ | International Financial Reporting Standards | Other ☐ |
| as issued by the International Accounting Standards Board ☒ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the Registrant has elected to follow: Item 17 ☐ Item 18 ☐
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
TABLE OF CONTENTS
i
USE OF CERTAIN TERMS
In this Annual Report on Form 20-F, unless the context otherwise requires:
| ● | references to “ADSs” refer to the Registrant’s American Depositary Shares; |
| ● | references to “A3AR” refer to the A3 adenosine receptor; |
| ● | references to the “Company,” “we,” “our” and “Can-fite” refer to Can-fite BioPharma Ltd. (the “Registrant”) and its consolidated subsidiaries; |
| ● | references to the “Companies Law” or “Israeli Companies Law” are to Israel’s Companies Law, 5759-1999, as amended; |
| ● | references to “dollars,” “U.S. dollars” and “$” are to United States Dollars; |
| ● | references to “HCC” refer to hepatocellular carcinoma, also known as primary liver cancer; |
| ● | references to “HCV” refer to hepatitis C virus; |
| ● | references to “ordinary shares,” “our shares” and similar expressions refer to the Registrant’s Ordinary Shares, NIS 0.25 nominal (par) value per share; | |
| ● | references to “NAFLD” refer to non-alcoholic fatty liver disease; | |
| ● | references to “NASH” refer to non-alcoholic steatohepatitis; |
| ● | references to “OA” refer to osteoarthritis; |
| ● | references to “PBMC” refer to peripheral blood mononuclear cells; |
| ● | references to “RA” refer to rheumatoid arthritis; |
| ● | references to “Securities Law” or “Israeli Securities Law” are to Israel Securities Law, 5728-1968, as amended; |
| ● | references to “shekels” and “NIS” are to New Israeli Shekels, the Israeli currency; and |
| ● | references to the “SEC” are to the United States Securities and Exchange Commission. |
ii
FORWARD LOOKING STATEMENTS
This Annual Report on Form 20-F contains forward-looking statements, about our expectations, beliefs or intentions regarding, among other things, our product development efforts, business, financial condition, results of operations, strategies or prospects. In addition, from time to time, we or our representatives have made or may make forward-looking statements, orally or in writing. Forward-looking statements can be identified by the use of forward-looking words such as “believe,” “expect,” “intend,” “plan,” “may,” “should” or “anticipate” or their negatives or other variations of these words or other comparable words or by the fact that these statements do not relate strictly to historical or current matters. These forward-looking statements may be included in, but are not limited to, various filings made by us with the U.S. Securities and Exchange Commission, or the SEC, press releases or oral statements made by or with the approval of one of our authorized executive officers. Forward-looking statements relate to anticipated or expected events, activities, trends or results as of the date they are made. Because forward-looking statements relate to matters that have not yet occurred, these statements are inherently subject to risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements. Many factors could cause our actual activities or results to differ materially from the activities and results anticipated in forward-looking statements, including, but not limited to, the factors summarized below.
This Annual Report on Form 20-F identifies important factors which could cause our actual results to differ materially from those indicated by the forward-looking statements, particularly those set forth under the heading “Risk Factors.” The risk factors included in this Annual Report on Form 20-F are not necessarily all of the important factors that could cause actual results to differ materially from those expressed in any of our forward-looking statements. Given these uncertainties, readers are cautioned not to place undue reliance on such forward-looking statements. Factors that could cause our actual results to differ materially from those expressed or implied in such forward-looking statements include, but are not limited to:
| ● | the initiation, timing, progress and results of our preclinical studies, clinical trials and other product candidate development efforts; |
| ● | our ability to advance our product candidates into clinical trials or to successfully complete our preclinical studies or clinical trials; |
| ● | our receipt of regulatory approvals for our product candidates, and the timing of other regulatory filings and approvals; |
| ● | the clinical development, commercialization and market acceptance of our product candidates; |
| ● | our ability to establish and maintain corporate collaborations; |
| ● | the implementation of our business model and strategic plans for our business and product candidates; |
| ● | the scope of protection we are able to establish and maintain for intellectual property rights covering our product candidates and our ability to operate our business without infringing the intellectual property rights of others; |
| ● | estimates of our expenses, future revenues, capital requirements and our needs for additional financing; |
| ● | competitive companies, technologies and our industry; and |
| ● | statements as to the impact of the political and security situation in Israel on our business. |
All forward-looking statements attributable to us or persons acting on our behalf speak only as of the date of this Annual Report on Form 20-F and are expressly qualified in their entirety by the cautionary statements included in this Annual Report on Form 20-F. We undertake no obligations to update or revise forward-looking statements to reflect events or circumstances that arise after the date made or to reflect the occurrence of unanticipated events. In evaluating forward-looking statements, you should consider these risks and uncertainties.
iii
EXPLANATORY NOTE
Market data and certain industry data and forecasts used throughout this Annual Report on Form 20-F were obtained from internal company surveys, market research, consultant surveys, publicly available information, reports of governmental agencies and industry publications and surveys. Industry surveys, publications, consultant surveys and forecasts generally state that the information contained therein has been obtained from sources believed to be reliable, but that the accuracy and completeness of such information is not guaranteed. We have not independently verified any of the data from third-party sources, nor have we ascertained the underlying economic assumptions relied upon therein. Similarly, internal surveys, industry forecasts and market research, which we believe to be reliable based upon our management’s knowledge of the industry, have not been independently verified. Forecasts are particularly likely to be inaccurate, especially over long periods of time. In addition, we do not necessarily know what assumptions regarding general economic growth were used in preparing the forecasts we cite. Statements as to our market position are based on the most currently available data. While we are not aware of any misstatements regarding the industry data presented in this Annual Report on Form 20-F, our estimates involve risks and uncertainties and are subject to change based on various factors, including those discussed under the heading “Risk Factors” in this Annual Report on Form 20-F.
iv
ITEM 1. Identity of Directors, Senior Management and Advisers.
Not applicable.
ITEM 2. Offer Statistics and Expected Timetable.
Not applicable.
A. Selected Financial Data.
The following table sets forth our selected consolidated financial data for the periods ended and as of the dates indicated. The following selected consolidated financial data for our company should be read in conjunction with the financial information, “Item 5. Operational and Financial Review and Prospects” and other information provided elsewhere in this Annual Report on Form 20-F and our consolidated financial statements and related notes. The selected consolidated financial data in this section is not intended to replace the consolidated financial statements and is qualified in its entirety thereby.
The selected consolidated statements of operations data for the years ended December 31, 2016, 2015 and 2014, and the selected consolidated balance sheet data as of December 31, 2016 and 2015, have been derived from our audited consolidated financial statements set forth elsewhere in this Annual Report on Form 20-F. The selected consolidated statements of operations data for the years ended December 31, 2013 and 2012, and the selected consolidated balance sheet data as of December 31, 2014, 2013 and 2012, have been derived from our audited consolidated financial statements not included in this Form 20-F.
Our consolidated financial statements included in this Annual Report on Form 20-F were prepared in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, and reported in Israeli New Shekels, or NIS.
| Consolidated Statements Of | Year Ended December 31, | |||||||||||||||||||||||
| Operations Data: | 2012 | 2013 | 2014 | 2015 | 2016 | 2016 | ||||||||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||||||||||
| NIS | Convenience translation to US $ | |||||||||||||||||||||||
| Revenues | - | - | - | 643 | 652 | 170 | ||||||||||||||||||
| Operating expenses: | ||||||||||||||||||||||||
| Research and development expenses, net | 13,160 | 15,390 | 16,200 | 15,052 | 23,380 | 6,081 | ||||||||||||||||||
| General and administrative expenses | 9,272 | 15,922 | 11,573 | 10,633 | 10,483 | 2,726 | ||||||||||||||||||
| Operating loss | 22,432 | 31,312 | 27,773 | 25,042 | 33,211 | 8,637 | ||||||||||||||||||
| Other expense – due to M&A | - | - | - | - | - | - | ||||||||||||||||||
| Financial expenses | 59 | 892 | 1,228 | 2,203 | 685 | 178 | ||||||||||||||||||
| Financial income | (573 | ) | (1401 | ) | (4,500 | ) | (7,492 | ) | (6,999 | ) | (1,820 | ) | ||||||||||||
| Taxes on income | 11 | 9 | 23 | 17 | 112 | 29 | ||||||||||||||||||
| Net loss | 21,929 | 30,812 | 24,524 | 19,770 | 27,009 | 7,024 | ||||||||||||||||||
| Adjustments arising from translating financial statements of foreign operations | (7 | ) | 206 | 939 | 1 | 33 | 9 | |||||||||||||||||
| Remeasurements loss (gain) from defined benefit plan | (42 | ) | 49 | 94 | 385 | - | - | |||||||||||||||||
| Comprehensive loss | 21,880 | 31,067 | 25, 557 | 20,156 | 27,042 | 7,033 | ||||||||||||||||||
| Net loss per ordinary share | 2.08 | 2.12 | 1.35 | 0.81 | 0.96 | 0.25 | ||||||||||||||||||
| Number of ordinary shares used in computing loss per ordinary share | 10,050,927 | 13,712,521 | 17,545,663 | 22,953,077 | 27,709,901 | - | ||||||||||||||||||
| 1 |
| Consolidated Balance | As of December 31, | |||||||||||||||||||||||
| Sheet Data: | 2012 | 2013 | 2014 | 2015 | 2016 | 2016 | ||||||||||||||||||
| (in thousands NIS) | (in thousands NIS) | (in thousands NIS) | (in thousands NIS) | (in thousands NIS) | (in US $ thousands) | |||||||||||||||||||
| Cash and cash equivalents | 4,278 | 20,767 | 36,091 | 66,026 | 31,203 | 8,115 | ||||||||||||||||||
| Other receivables and lease deposit | 1,672 | 2,195 | 3,443 | 2,446 | 7,701 | 2,003 | ||||||||||||||||||
| Fixed assets | 159 | 143 | 133 | 236 | 205 | 53 | ||||||||||||||||||
| Total assets | 6,109 | 23,105 | 39,667 | 68,708 | 39,109 | 10,171 | ||||||||||||||||||
| Total liabilities | 8,754 | 7,580 | 12,967 | 27,935 | 24,207 | 6,295 | ||||||||||||||||||
| Total shareholders’ equity | (2,645 | ) | 15,525 | 26,700 | 40,773 | 14,902 | 3,876 | |||||||||||||||||
We report our financial statements in NIS. This Annual Report on Form 20-F contains conversions of NIS amounts into U.S. dollars at specific rates solely for the convenience of the reader. Unless otherwise noted, for the purposes of annual financial data, all conversions from NIS to U.S. dollars and from U.S. dollars to NIS were made at a rate of NIS 3.902 to $1.00 U.S. dollar, the daily representative rates in effect as of December 31, 2016. No representation is made that the NIS amounts referred to in this Annual Report on Form 20-F could have been or could be converted into U.S. dollars at any particular rate or at all.
The following table sets forth information regarding the exchange rates of U.S. dollars per NIS for the periods indicated. Average rates are calculated by using the daily representative rates as reported by the Bank of Israel on the last day of each month during the periods presented.
| NIS per U.S. $ | ||||||||||||||||
| Year Ended December 31, | High | Low | Average | Period End | ||||||||||||
| 2016 | 3.983 | 3.746 | 3.841 | 3.845 | ||||||||||||
| 2015 | 4.053 | 3.761 | 3.887 | 3.902 | ||||||||||||
| 2014 | 3.994 | 3.402 | 3.578 | 3.889 | ||||||||||||
| 2013 | 3.791 | 3.471 | 3.611 | 3.471 | ||||||||||||
| 2012 | 4.084 | 3.700 | 3.858 | 3.733 | ||||||||||||
The following table sets forth the high and low daily representative rates for the NIS as reported by the Bank of Israel for each of the prior six months.
| NIS per U.S. $ | ||||||||||||||||
| Month Ended | High | Low | Average | Period End | ||||||||||||
| March 2017 (through March 29, 2017) | 3.693 | 3.614 | 3.655 | 3.625 | ||||||||||||
| February 2017 | 3.768 | 3.659 | 3.763 | 3.659 | ||||||||||||
| January 2017 | 3.860 | 3.769 | 3.818 | 3.769 | ||||||||||||
| December 2016 | 3.867 | 3.787 | 3.829 | 3.845 | ||||||||||||
| November 2016 | 3.876 | 3.799 | 3.843 | 3.839 | ||||||||||||
| October 2016 | 3.856 | 3.778 | 3.822 | 3.849 | ||||||||||||
| September 2016 | 3.786 | 3.746 | 3.766 | 3.758 | ||||||||||||
On March 29, 2017, the closing representative rate was $1.00 to NIS 3.625, as reported by the Bank of Israel.
| 2 |
B. Capitalization and Indebtedness.
Not applicable.
C. Reasons for the Offer and Use of Proceeds.
Not applicable.
D. Risk Factors
You should carefully consider the risks we describe below, in addition to the other information set forth elsewhere in this Annual Report on Form 20-F, including our consolidated financial statements and the related notes beginning on page F-1, before deciding to invest in our ordinary shares and ADSs. These material risks could adversely impact our results of operations, possibly causing the trading price of our ordinary shares and ADSs to decline, and you could lose all or part of your investment.
Risks Related to Our Financial Position and Capital Requirements
We have incurred operating losses since our inception and anticipate that we will continue to incur substantial operating losses for the foreseeable future.
We are a clinical stage biopharmaceutical company that develops orally bioavailable small molecule therapeutic products for the treatment of autoimmune-inflammatory indications, oncology and liver diseases as well as sexual dysfunction. Since our incorporation in 1994, we have been focused on research and development activities with a view to developing our product candidates, CF101also known as Piclidenoson, CF102, also known as Namodenoson, and CF602. We have financed our operations primarily through the sale of equity securities (both in private placements and in public offerings on the Tel Aviv Stock Exchange, or TASE and NYSE MKT) and payments received under out-licensing agreements and have incurred losses in each year since our inception in 1994. We have historically incurred substantial net losses, including net losses of approximately NIS 27 million (approximately $7 million) in 2016, NIS 20 million (approximately $5 million) in 2015 and NIS 25 million in 2014 (approximately $6 million). At December 31, 2016, we had an accumulated deficit of approximately NIS 350 million (approximately $91 million). We do not know whether or when we will become profitable. To date, we have not commercialized any products or generated any revenues from product sales and accordingly we do not have a revenue stream to support our cost structure. Our losses have resulted principally from costs incurred in development and discovery activities. We expect to continue to incur losses for the foreseeable future, and these losses will likely increase as we:
| ● | initiate and manage pre-clinical development and clinical trials for our current and new product candidates; |
| ● | seek regulatory approvals for our product candidates; |
| ● | implement internal systems and infrastructures; |
| ● | seek to license additional technologies to develop; |
| ● | hire management and other personnel; and |
| ● | move towards commercialization. |
| 3 |
If our product candidates fail in clinical trials or do not gain regulatory clearance or approval, or if our product candidates do not achieve market acceptance, we may never become profitable. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our inability to achieve and then maintain profitability would negatively affect our business, financial condition, results of operations and cash flows. Moreover, our prospects must be considered in light of the risks and uncertainties encountered by an early-stage company and in highly regulated and competitive markets, such as the biopharmaceutical market, where regulatory approval and market acceptance of our products are uncertain. There can be no assurance that our efforts will ultimately be successful or result in revenues or profits.
We will need to raise additional capital to meet our business requirements in the future, and such capital raising may be costly or difficult to obtain and will dilute current shareholders’ ownership interests.
As of December 31, 2016, we had cash and cash equivalents of approximately NIS 31 million (approximately $8 million). During the fourth quarter of 2016, we received approximately NIS 1.9 million ($0.5 million) from Chong Kun Dang Pharmaceuticals, or CKD, as upfront payment for entering into the distribution agreement with CKD and in January 2017, we raised approximately NIS 18.9 million (approximately $5 million) in a registered direct offering. We believe that our existing financial resources will be sufficient to meet our requirements for the next twelve months. We have expended and believe that we will continue to expend substantial resources for the foreseeable future developing our product candidates. These expenditures will include costs associated with research and development, manufacturing, conducting preclinical experiments and clinical trials and obtaining regulatory approvals, as well as commercializing any products approved for sale. Because the outcome of our planned and anticipated clinical trials is highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of our product candidates. In addition, other unanticipated costs may arise. As a result of these and other factors currently unknown to us, we will require additional funds, through public or private equity or debt financings or other sources, such as strategic partnerships and alliances and licensing arrangements. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
Our future capital requirements will depend on many factors, including the progress and results of our clinical trials, the duration and cost of discovery and preclinical development, and laboratory testing and clinical trials for our product candidates, the timing and outcome of regulatory review of our product candidates, the number and development requirements of other product candidates that we pursue, and the costs of activities, such as product marketing, sales, and distribution. Because of the numerous risks and uncertainties associated with the development and commercialization of our product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our anticipated clinical trials.
Our future capital requirements depend on many factors, including:
●
● |
the level of research and development investment required to develop our product candidates;
the failure to obtain regulatory approval or achieve commercial success of our product candidates, including Piclidenoson, Namodenoson and CF602; |
| ● | the results of our preclinical studies and clinical trials for our earlier stage product candidates, and any decisions to initiate clinical trials if supported by the preclinical results; |
| ● | the costs, timing and outcome of regulatory review of our product candidates that progress to clinical trials; |
| ● | the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our issued patents and defending intellectual property-related claims; |
| ● | the cost of commercialization activities if any of our product candidates are approved for sale, including marketing, sales and distribution costs; |
| ● | the cost of manufacturing our product candidates and any products we successfully commercialize; |
| ● | the timing, receipt and amount of sales of, or royalties on, our future products, if any; |
| ● | the expenses needed to attract and retain skilled personnel; |
| 4 |
| ● | any product liability or other lawsuits related to our products; |
| ● | the extent to which we acquire or invest in businesses, products or technologies and other strategic relationships; |
● |
the costs of financing unanticipated working capital requirements and responding to competitive pressures; and | |
| ● | maintaining minimum shareholders’ equity requirements under the NYSE MKT Company Guide. |
Additional funds may not be available when we need them, on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to delay, limit, reduce or terminate preclinical studies, clinical trials or other research and development activities for one or more of our product candidates or delay, limit, reduce or terminate our establishment of sales and marketing capabilities or other activities that may be necessary to commercialize our product candidates.
We may incur substantial costs in pursuing future capital financing, including investment banking fees, legal fees, accounting fees, securities law compliance fees, printing and distribution expenses and other costs. We may also be required to recognize non-cash expenses in connection with certain securities we issue, such as convertible notes and warrants, which may adversely impact our financial condition.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
We may seek additional capital through a combination of private and public equity offerings, debt financings, strategic partnerships and alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of existing shareholders will be diluted, and the terms may include liquidation or other preferences that adversely affect shareholder rights. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take certain actions, such as incurring debt, making capital expenditures or declaring dividends. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us. If we are unable to raise additional funds through equity or debt financing when needed, we may be required to delay, limit, reduce or terminate our product development or commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Risks Related to our Business and Regulatory Matters
We have not yet commercialized any products or technologies, and we may never become profitable.
We have not yet commercialized any products or technologies, and we may never be able to do so. We do not know when or if we will complete any of our product development efforts, obtain regulatory approval for any product candidates incorporating our technologies or successfully commercialize any approved products. Even if we are successful in developing products that are approved for marketing, we will not be successful unless these products gain market acceptance for appropriate indications at favorable reimbursement rates. The degree of market acceptance of these products will depend on a number of factors, including:
| ● | the timing of regulatory approvals in the countries, and for the uses, we seek; |
| ● | the competitive environment; |
| ● | the establishment and demonstration in the medical community of the safety and clinical efficacy of our products and their potential advantages over existing therapeutic products; |
| ● | our ability to enter into strategic agreements with pharmaceutical and biotechnology companies with strong marketing and sales capabilities; |
| ● | the adequacy and success of distribution, sales and marketing efforts; and |
| ● | the pricing and reimbursement policies of government and third-party payors, such as insurance companies, health maintenance organizations and other plan administrators. |
| 5 |
Physicians, patients, thirty-party payors or the medical community in general may be unwilling to accept, utilize or recommend, and in the case of third-party payors, cover any of our products or products incorporating our technologies. As a result, we are unable to predict the extent of future losses or the time required to achieve profitability, if at all. Even if we successfully develop one or more products that incorporate our technologies, we may not become profitable.
Our product candidates are at various stages of clinical and preclinical development and may never be commercialized.
Our product candidates are at various stages of clinical development and may never be commercialized. The progress and results of any future pre-clinical testing or future clinical trials are uncertain, and the failure of our product candidates to receive regulatory approvals will have a material adverse effect on our business, operating results and financial condition to the extent we are unable to commercialize any products. None of our product candidates has received regulatory approval for commercial sale. In addition, we face the risks of failure inherent in developing therapeutic products. Our product candidates are not expected to be commercially available for several years, if at all.
In addition, our product candidates must satisfy rigorous standards of safety and efficacy before they can be approved by the U.S. Food and Drug Administration, or the FDA, the European Medicines Agency, or EMA, and foreign regulatory authorities for commercial use. The FDA, EMA and foreign regulatory authorities have full discretion over this approval process. We will need to conduct significant additional research, involving testing in animals and in humans, before we can file applications for product approval. Typically, in the pharmaceutical industry, there is a high rate of attrition for product candidates in pre-clinical testing and clinical trials. Also, satisfying regulatory requirements typically takes many years, is dependent upon the type, complexity and novelty of the product and requires the expenditure of substantial resources. In addition, delays or rejections may be encountered based upon additional government regulation, including any changes in FDA policy, during the process of product development, clinical trials and regulatory reviews.
In order to receive FDA approval or approval from foreign regulatory authorities to market a product candidate or to distribute our products, we must demonstrate thorough pre-clinical testing and thorough human clinical trials that the product candidate is safe and effective for its intended uses (e.g., treatment of a specific condition in a specific way subject to contradictions and other limitations). Even if we comply with all FDA requests, the FDA may ultimately reject one or more of our new drug applications, or NDA, or grant approval for a narrowly intended use that is not commercially feasible. We might not obtain regulatory approval for our drug candidates in a timely manner, if at all. Failure to obtain FDA approval of any of our drug candidates in a timely manner or at all will severely undermine our business by reducing the number of salable products and, therefore, corresponding product revenues.
Results of earlier clinical trials may not be predictive of the results of later-stage clinical trials.
The results of preclinical studies and early clinical trials of product candidates may not be predictive of the results of later-stage clinical trials. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy results despite having progressed through preclinical studies and initial clinical trials. For example, in December 2013, OphthaliX, our subsidiary, announced top-line results of a Phase III study with Piclidenoson for dry-eye syndrome in which Piclidenoson did not meet the primary efficacy endpoint of complete clearing of corneal staining, nor the secondary efficacy endpoints and in July 2016, OphthaliX released top-line results from its Phase II clinical trial of Piclidenoson for the treatment of glaucoma in which no statistically significant differences were found between the Piclidenoson treated group and the placebo group in the primary endpoint of lowering intra ocular pressure. In addition, two Phase IIb studies in rheumatoid arthritis, or RA, utilizing Piclideneson in combination with methotrexate, a generic drug commonly used for treating RA patients, or MTX, failed to reach their primary endpoints. We believe that this may have been due to low A3AR expression in the subpopulation of RA patients that did not respond well to treatment with MTX. Because of their low A3AR expression, such patients also did not respond well to treatment with Piclidenoson. We were not aware of this when we designed the studies. As such, we conducted an additional Phase IIb RA trial of Piclidenoson as a standalone therapy in patients with A3AR expression levels above a certain threshold, and positive results from this study were announced in December 2013. Furthermore, a Phase II/III study of Piclideneson for psoriasis did not meet its primary endpoint although positive data from further analysis of the Phase II/III study suggests Piclidenoson as a potential systemic therapy for patients with moderate-severe psoriasis.
| 6 |
Many companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials due to adverse safety profiles or lack of efficacy, notwithstanding promising results in earlier studies. Any delay in, or termination or suspension of, our clinical trials will delay the requisite filings with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues. If the clinical trials do not support our product claims, the completion of development of such product candidates may be significantly delayed or abandoned, which will significantly impair our ability to generate product revenues and will materially adversely affect our results of operations.
This drug candidate development risk is heightened by any changes in the planned clinical trials compared to the completed clinical trials. As product candidates are developed from preclinical through early to late stage clinical trials towards approval and commercialization, it is customary that various aspects of the development program, such as manufacturing and methods of administration, are altered along the way in an effort to optimize processes and results. While these types of changes are common and are intended to optimize the product candidates for late stage clinical trials, approval and commercialization, such changes do carry the risk that they will not achieve these intended objectives.
Changes in our planned clinical trials or future clinical trials could cause our product candidates to perform differently, including causing toxicities, which could delay completion of our clinical trials, delay approval of our product candidates, and/or jeopardize our ability to commence product sales and generate revenues.
We might be unable to develop product candidates that will achieve commercial success in a timely and cost-effective manner, or ever.
Even if regulatory authorities approve our product candidates, they may not be commercially successful. Our product candidates may not be commercially successful because government agencies and other third-party payors may not cover the product or the coverage may be too limited to be commercially successful; physicians and others may not use or recommend our products, even following regulatory approval. A product approval, assuming one issues, may limit the uses for which the product may be distributed thereby adversely affecting the commercial viability of the product. Third parties may develop superior products or have proprietary rights that preclude us from marketing our products. We also expect that at least some of our product candidates will be expensive, if approved. Patient acceptance of and demand for any product candidates for which we obtain regulatory approval or license will depend largely on many factors, including but not limited to the extent, if any, of reimbursement of costs by government agencies and other third-party payors, pricing, the effectiveness of our marketing and distribution efforts, the safety and effectiveness of alternative products, and the prevalence and severity of side effects associated with our products. If physicians, government agencies and other third-party payors do not accept our products, we will not be able to generate significant revenue.
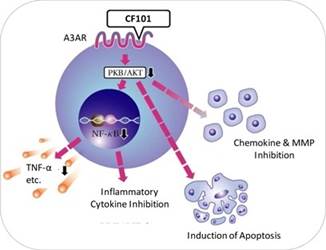
Our current pipeline is based on our platform technology utilizing the Gi protein associated A3 adenosine receptor, or A3AR, as a potent therapeutic target and currently includes three molecules, Piclidenoson, Namodenoson and CF602 product candidates, of which Piclidenoson is the most advanced. Failure to develop these molecules will have a material adverse effect on us.
Our current pipeline is based on a platform technology where we target the A3AR with highly selective ligands, or small signal triggering molecules that bind to specific cell surface receptors, such as the A3AR, including Piclidenoson, Namodenoson and CF602. A3ARs are structures found in cell surfaces that record and transfer messages from small molecules or ligands, such as Piclidenoson, Namodenoson and CF602 to the rest of the cell. Piclidenoson is the most advanced of our drug candidates. As such, we are currently dependent on only three molecules for our potential commercial success, and any safety or efficacy concerns related to such molecules would have a significant impact on our business. Failure to develop our drug candidates, in whole or in part, will have a material adverse effect on us.
Clinical trials are very expensive, time-consuming and difficult to design and implement, and, as a result, we may suffer delays or suspensions in future trials which would have a material adverse effect on our ability to generate revenues.
Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. Regulatory authorities, such as the FDA, may preclude clinical trials from proceeding. Additionally, the clinical trial process is time-consuming, failure can occur at any stage of the trials, and we may encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed by several factors, including:
| ● | unforeseen safety issues; |
| ● | determination of dosing issues; |
| 7 |
| ● | lack of effectiveness or efficacy during clinical trials; |
| ● | failure of third party suppliers to perform final manufacturing steps for the drug substance; |
| ● | slower than expected rates of patient recruitment and enrollment; |
| ● | lack of healthy volunteers and patients to conduct trials; |
| ● | inability to monitor patients adequately during or after treatment; |
| ● | failure of third party contract research organizations to properly implement or monitor the clinical trial protocols; |
| ● | failure of institutional review boards to approve our clinical trial protocols; |
| ● | inability or unwillingness of medical investigators and institutional review boards to follow our clinical trial protocols; and |
| ● | lack of sufficient funding to finance the clinical trials. |
We have experienced the risks involved with conducting clinical trials, including but not limited to, increased expense and delay and failure to meet end points of the trial. For example, in December 2013, OphthaliX, our subsidiary, announced top-line results of a Phase III study with CF 101 for dry-eye syndrome in which Piclidenoson did not meet the primary efficacy endpoint of complete clearing of corneal staining, nor the secondary efficacy endpoints and in July 2016, OphthaliX released top-line results from its Phase II clinical trial of Piclidenoson for the treatment of glaucoma in which no statistically significant differences were found between the Piclidenoson treated group and the placebo group in the primary endpoint of lowering intra ocular pressure. In addition, two Phase IIb studies in RA, utilizing Piclidenoson in combination with methotrexate, a generic drug commonly used for treating RA patients, or MTX, failed to reach their primary end points. We believe that this may have been due to low A3AR expression in the subpopulation of RA patients that did not respond well to treatment with MTX. Because of their low A3AR expression, such patients also did not respond well to treatment with Piclidenoson. We were not aware of this when it designed the studies. As such, we conducted an additional Phase IIb RA trial of Piclidenoson as a standalone therapy in patients with A3AR expression levels above a certain threshold, and positive results from this study were announced in December 2013. Furthermore, a Phase II/III study of Piclidenoson for psoriasis did not meet its primary endpoint although positive data from further analysis of the Phase II/III study suggests Piclidenoson as a potential systemic therapy for patients with moderate-severe psoriasis.
In addition, we or regulatory authorities may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the regulatory authorities find deficiencies in our regulatory submissions or the conduct of these trials. Any suspension of clinical trials will delay possible regulatory approval, if any, and adversely impact our ability to develop products and generate revenue.
If we acquire or license additional technology or product candidates, we may incur a number of costs, may have integration difficulties and may experience other risks that could harm our business and results of operations.
We may acquire and license additional product candidates and technologies. Any product candidate or technology we license from others or acquire will likely require additional development efforts prior to commercial sale, including extensive pre-clinical or clinical testing, or both, and approval by the FDA and applicable foreign regulatory authorities, if any. All product candidates are prone to risks of failure inherent in pharmaceutical product development, including the possibility that the product candidate or product developed based on licensed technology will not be shown to be sufficiently safe and effective for approval by regulatory authorities. In addition, we cannot assure you that any product candidate that we develop based on acquired or licensed technology that is granted regulatory approval will be manufactured or produced economically, successfully commercialized or widely accepted in the marketplace. Moreover, integrating any newly acquired product candidates could be expensive and time-consuming. If we cannot effectively manage these aspects of our business strategy, our business may not succeed.
| 8 |
The manufacture of our product candidates is a chemical synthesis process and if one of our materials suppliers encounters problems manufacturing our products, our business could suffer.
The FDA and foreign regulators require manufacturers to register manufacturing facilities. The FDA and foreign regulators also inspect these facilities to confirm compliance with requirements that the FDA or foreign regulators establish. We do not intend to engage in the manufacture of our products other than for pre-clinical and clinical studies, but we or our materials suppliers may face manufacturing or quality control problems causing product production and shipment delays or a situation where we or the supplier may not be able to maintain compliance with the FDA’s or foreign regulators’ requirements necessary to continue manufacturing our drug substance. Drug manufacturers are subject to ongoing periodic unannounced inspections by the FDA, the U.S. Drug Enforcement Agency, or DEA, and corresponding foreign regulators to ensure strict compliance with requirements and other governmental regulations and corresponding foreign standards. Any failure to comply with DEA requirements or FDA or foreign regulatory requirements could adversely affect our clinical research activities and our ability to market and develop our product candidates.
We do not currently have sales, marketing or distribution capabilities or experience, and we are unable to effectively sell, market or distribute our product candidates now and we do not expect to be able to do so in the future. The failure to enter into agreements with third parties that are capable of performing these functions would have a material adverse effect on our business and results of operations.
We do not currently have and we do not expect to develop sales, marketing and distribution capabilities. If we are unable to enter into agreements with third parties to perform these functions, we will not be able to successfully market any of our platforms or product candidates. In order to successfully market any of our platform or product candidates, we must make arrangements with third parties to perform these services.
As we do not intend to develop a marketing and sales force with technical expertise and supporting distribution capabilities, we will be unable to market any of our product candidates directly. To promote any of our potential products through third parties, we will have to locate acceptable third parties for these functions and enter into agreements with them on acceptable terms, and we may not be able to do so. Any third-party arrangements we are able to enter into may result in lower revenues than we could achieve by directly marketing and selling our potential products. In addition, to the extent that we depend on third parties for marketing and distribution, any revenues we receive will depend upon the efforts of such third parties, as well as the terms of our agreements with such third parties, which cannot be predicted in most cases at this time. As a result, we might not be able to market and sell our products in the United States or overseas, which would have a material adverse effect on us.
We will to some extent rely on third parties to implement our manufacturing and supply strategies. Failure of these third parties in any respect could have a material adverse effect on our business, results of operations and financial condition.
If our current and future manufacturing and supply strategies are unsuccessful, then we may be unable to conduct and complete any future pre-clinical or clinical trials or commercialize our product candidates in a timely manner, if at all. Completion of any potential future pre-clinical or clinical trials and commercialization of our product candidates will require access to, or development of, facilities to manufacture a sufficient supply of our product candidates. We do not have the resources, facilities or experience to manufacture our product candidates for commercial purposes on our own and do not intend to develop or acquire facilities for the manufacture of product candidates for commercial purposes in the foreseeable future. We may rely on contract manufacturers to produce sufficient quantities of our product candidates necessary for any pre-clinical or clinical testing we undertake in the future. Such contract manufacturers may be the sole source of production and they may have limited experience at manufacturing, formulating, analyzing, filling and finishing our types of product candidates.
We also intend to rely on third parties to supply the requisite materials needed for the manufacturing of our active pharmaceutical ingredients, or API. There may be a limited supply of these requisite materials. We might not be able to enter into agreements that provide us assurance of availability of such components in the future from any supplier. Our potential suppliers may not be able to adequately supply us with the components necessary to successfully conduct our pre-clinical and clinical trials or to commercialize our product candidates. If we cannot acquire an acceptable supply of the requisite materials to produce our product candidates, we will not be able to complete pre-clinical and clinical trials and will not be able to market or commercialize our product candidates.
| 9 |
We depend on key members of our management and key consultants and will need to add and retain additional leading experts. Failure to retain our management and consulting team and add additional leading experts could have a material adverse effect on our business, results of operations or financial condition.
We are highly dependent on our executive officers and other key management and technical personnel. Our failure to retain our Chief Executive Officer, Pnina Fishman, Ph.D., who has developed much of the technology we utilize today, or any other key management and technical personnel, could have a material adverse effect on our future operations. Our success is also dependent on our ability to attract, retain and motivate highly trained technical, and management personnel, among others, to continue the development and commercialization of our current and future products. We presently maintain a life insurance policy on our Chief Executive Officer, Pnina Fishman.
Our success also depends on our ability to attract, retain and motivate personnel required for the development, maintenance and expansion of our activities. There can be no assurance that we will be able to retain our existing personnel or attract additional qualified employees or consultants. The loss of key personnel or the inability to hire and retain additional qualified personnel in the future could have a material adverse effect on our business, financial condition and results of operation.
We face significant competition and continuous technological change, and developments by competitors may render our products or technologies obsolete or non-competitive. If we cannot successfully compete with new or existing products, our marketing and sales will suffer and we may not ever be profitable.
We will compete against fully integrated pharmaceutical and biotechnology companies and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. In addition, many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs than we do, and have substantially greater financial resources than we do, as well as significantly greater experience in:
| ● | developing drugs; |
| ● | undertaking pre-clinical testing and human clinical trials; |
| ● | obtaining FDA, addressing various regulatory matters and other regulatory approvals of drugs; |
| ● | formulating and manufacturing drugs; and |
| ● | launching, marketing and selling drugs. |
If our competitors develop and commercialize products faster than we do, or develop and commercialize products that are superior to our product candidates, our commercial opportunities will be reduced or eliminated. The extent to which any of our product candidates achieve market acceptance will depend on competitive factors, many of which are beyond our control. Competition in the biotechnology and biopharmaceutical industry is intense and has been accentuated by the rapid pace of technology development. Our competitors include large integrated pharmaceutical companies, biotechnology companies that currently have drug and target discovery efforts, universities, and public and private research institutions. Almost all of these entities have substantially greater research and development capabilities and financial, scientific, manufacturing, marketing and sales resources than we do. These organizations also compete with us to:
| ● | attract parties for acquisitions, joint ventures or other collaborations; |
| ● | license proprietary technology that is competitive with the technology we are developing; |
| ● | attract funding; and |
| ● | attract and hire scientific talent and other qualified personnel. |
Our competitors may succeed in developing and commercializing products earlier and obtaining regulatory approvals from the FDA or foreign regulators more rapidly than we do. Our competitors may also develop products or technologies that are superior to those we are developing, and render our product candidates or technologies obsolete or non-competitive. If we cannot successfully compete with new or existing products, our marketing and sales will suffer and we may not ever be profitable.
| 10 |
Our competitors currently include companies with marketed products and/or an advanced research and development pipeline. The major competitors in the arthritis and psoriasis therapeutic field include Amgen, Centocor, Pfizer, Novartis, Abbvie, Celgene, Eli Lilly, Janssen and more. Competitors in the hepatocellular carcinoma, also known as primary liver cancer, or HCC field include companies such as Bayer. Competitors in the NASH field include companies such as Gilead, Genfit, Regato, Galmed, Raptor and Intercept. Competitors in the erectile dysfunction field include Pfizer, Eli Lilly and Bayer. See “Item 4. Information on the Company—B. Business Overview—Competition.”
Moreover, several companies have reported the commencement of research projects related to the A3AR. Such companies include CV Therapeutics Inc. (which was acquired by Gilead), King Pharmaceuticals R&D Inv. (which was acquired by Pfizer), Hoechst Marion Roussel Inc. (which was acquired by Aventis), Novo Nordisk A/S and Inotek Pharmaceuticals. However, to the best of our knowledge, there is no approved drug currently on the market which is similar to our A3AR agonists, nor are we aware of any allosteric modulatorin the A3AR product pipeline similar to our allosteric modulator with respect to chemical profile and mechanism of action.
We are subject to a purported class action lawsuit. This litigation could result in substantial damages and may divert management’s time and attention from our business.
On June 29, 2015, we received a lawsuit, filed with the District Court of Tel-Aviv, requesting recognition of this lawsuit as a class action. The lawsuit named the Company, its Chief Executive Officer and directors as defendants. The lawsuit alleges, among other things, that we misled the public with regard to disclosures concerning the efficacy of our drug candidate, Piclidenoson. The claimant alleges that he suffered personal damages of over NIS 73,000, while also claiming that our shareholders suffered damages of approximately NIS 125 million. On March 31, 2016, we filed a response to the lawsuit. On March 1, 2017, a hearing was held in the District Court on whether to certify the lawsuit as a class action. A final hearing on the certification is scheduled for April 26, 2017. While we strongly believe the lawsuit and its allegations to be baseless and without merit and will vigorously defend this action, due to the inherent uncertainties that accompany litigation of this nature, there can be no assurance that we will be successful, and an adverse resolution of the lawsuit may result in damages beyond our insurance coverage for such cases, which could cause a risk of loss and expenditures that may adversely affect our financial condition and results of operations. Furthermore, this action may divert management’s time and attention from our business, and we could be forced to expend significant resources and pay significant costs and expenses, including legal fees, in connection with defending this lawsuit.
We may suffer losses from product liability claims if our product candidates cause harm to patients.
Any of our product candidates could cause adverse events. Although data from a pooled analysis of approximately 1,200 patients with inflammatory disease treated with Piclidenoson, indicates that Piclidenoson is safe and well tolerated at doses up to 4.0 mg administered twice daily for up to 12-32 weeks, there were incidences (albeit less than or equal to 5%) of adverse events in eight completed and fully analyzed trials in inflammatory disease. Such adverse events included nausea, diarrhea, constipation, common bacterial and viral syndromes (such as tonsillitis, otitis and respiratory and urinary tract infections), myalgia, arthralgia, dizziness, headache, palpitations and pruritus. We observed an even lower incidence (less than or equal to 2%) of serious adverse events, although only one type of event was reported in more than a single CF101-treated subject, which was exacerbation of chronic obstructive lung disease reported in two subjects. Notwithstanding the foregoing, the placebo group in such studies had a higher incidence of overall adverse events than the pooled Piclidenoson groups. No new safety concerns have been identified and no novel or unexpected safety concerns have appeared over 32 weeks of treatment in more recent trials.
There is also a risk that certain adverse events may not be observed in clinical trials, but may nonetheless occur in the future. If any of these adverse events occur, they may render our product candidates ineffective or harmful in some patients, and our sales would suffer, materially adversely affecting our business, financial condition and results of operations.
In addition, potential adverse events caused by our product candidates could lead to product liability lawsuits. If product liability lawsuits are successfully brought against us, we may incur substantial liabilities and may be required to limit the marketing and commercialization of our product candidates. Our business exposes us to potential product liability risks, which are inherent in the testing, manufacturing, marketing and sale of pharmaceutical products. We may not be able to avoid product liability claims. Product liability insurance for the pharmaceutical and biotechnology industries is generally expensive, if available at all. If, at any time, we are unable to obtain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims, we may be unable to clinically test, market or commercialize our product candidates. A successful product liability claim brought against us in excess of our insurance coverage, if any, may cause us to incur substantial liabilities, and, as a result, our business, liquidity and results of operations would be materially adversely affected.
| 11 |
Our product candidates will remain subject to ongoing regulatory requirements even if they receive marketing approval, and if we fail to comply with these requirements, we could lose these approvals, and the sales of any approved commercial products could be suspended.
Even if we receive regulatory approval to market a particular product candidate, the product will remain subject to extensive regulatory requirements, including requirements relating to manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, distribution and recordkeeping. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the uses for which the product may be marketed or the conditions of approval, or may contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product, which could negatively impact us or our collaboration partners by reducing revenues or increasing expenses, and cause the approved product candidate not to be commercially viable. In addition, as clinical experience with a drug expands after approval, typically because it is used by a greater number and more diverse group of patients after approval than during clinical trials, side effects and other problems may be observed after approval that were not seen or anticipated during pre-approval clinical trials or other studies. Any adverse effects observed after the approval and marketing of a product candidate could result in limitations on the use of or withdrawal of any approved products from the marketplace. Absence of long-term safety data may also limit the approved uses of our products, if any. If we fail to comply with the regulatory requirements of the FDA and other applicable U.S. and foreign regulatory authorities, or previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks, including the following:
| ● | Restrictions on the products, manufacturers or manufacturing process; |
| ● | Warning letters; |
| ● | Civil or criminal penalties, fines and injunctions; |
| ● | Product seizures or detentions; |
| ● | Import or export bans or restrictions; |
| ● | Voluntary or mandatory product recalls and related publicity requirements; |
| ● | Suspension or withdrawal of regulatory approvals; |
| ● | Total or partial suspension of production, and |
| ● | Refusal to approve pending applications for marketing approval of new products or supplements to approved applications. |
If we or our collaborators are slow or unable to adapt to changes in existing regulatory requirements or adoption of new regulatory requirements or policies, marketing approval for our product candidates may be lost or cease to be achievable, resulting in decreased revenue from milestones, product sales or royalties, which would have a material adverse effect on our results of operations.
We deal with hazardous materials and must comply with environmental, health and safety laws and regulations, which can be expensive and restrict how we do business.
Our activities and those of our third-party manufacturers on our behalf involve the controlled storage, use and disposal of hazardous materials, including corrosive, explosive and flammable chemicals and other hazardous compounds. We and our manufacturers are subject to U.S. federal, state, local, Israeli and other foreign laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental contamination or injury from these materials. In addition, if we develop a manufacturing capacity, we may incur substantial costs to comply with environmental regulations and would be subject to the risk of accidental contamination or injury from the use of hazardous materials in our manufacturing process.
| 12 |
In the event of an accident, government authorities may curtail our use of these materials and interrupt our business operations. In addition, we could be liable for any civil damages that result, which may exceed our financial resources and may seriously harm our business. Although our Israeli insurance program covers certain unforeseen sudden pollutions, we do not maintain a separate insurance policy for any of the foregoing types of risks. In addition, although the general liability section of our life sciences policy covers certain unforeseen, sudden environmental issues, pollution in the United States and Canada is excluded from the policy. In the event of environmental discharge or contamination or an accident, we may be held liable for any resulting damages, and any liability could exceed our resources. In addition, we may be subject to liability and may be required to comply with new or existing environmental laws regulating pharmaceuticals or other medical products in the environment.
We rely significantly on information technology and any failure, inadequacy, interruption or security lapse of that technology, including any cybersecurity incidents, could harm our ability to operate our business effectively.
Despite the implementation of security measures, our internal computer systems and those of third parties with which we contract are vulnerable to damage from cyber-attacks, computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. System failures, accidents or security breaches could cause interruptions in our operations, and could result in a material disruption of our clinical activities and business operations, in addition to possibly requiring substantial expenditures of resources to remedy. The loss of clinical trial data could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and our research and development programs and the development of our product candidates could be delayed.
We may not be able to successfully grow and expand our business. Failure to manage our growth effectively will have a material adverse effect on our business, results of operations and financial condition.
We may not be able to successfully grow and expand. Successful implementation of our business plan will require management of growth, including potentially rapid and substantial growth, which will result in an increase in the level of responsibility for management personnel and place a strain on our human and capital resources. To manage growth effectively, we will be required to continue to implement and improve our operating and financial systems and controls to expand, train and manage our employee base. Our ability to manage our operations and growth effectively requires us to continue to expend funds to enhance our operational, financial and management controls, reporting systems and procedures and to attract and retain sufficient numbers of talented personnel. If we are unable to scale up and implement improvements to our control systems in an efficient or timely manner, or if we encounter deficiencies in existing systems and controls, then we will not be able to make available the products required to successfully commercialize our technology. Failure to attract and retain sufficient numbers of talented personnel will further strain our human resources and could impede our growth or result in ineffective growth. Moreover, the management, systems and controls currently in place or to be implemented may not be adequate for such growth, and the steps taken to hire personnel and to improve such systems and controls might not be sufficient. If we are unable to manage our growth effectively, it will have a material adverse effect on our business, results of operations and financial condition.
If we are unable to obtain adequate insurance, our financial condition could be adversely affected in the event of uninsured or inadequately insured loss or damage. Our ability to effectively recruit and retain qualified officers and directors could also be adversely affected if we experience difficulty in obtaining adequate directors’ and officers’ liability insurance.
We may not be able to obtain insurance policies on terms affordable to us that would adequately insure our business and property against damage, loss or claims by third parties. To the extent our business or property suffers any damages, losses or claims by third parties, which are not covered or adequately covered by insurance, our financial condition may be materially adversely affected.
We may be unable to maintain sufficient insurance as a public company to cover liability claims made against our officers and directors. If we are unable to adequately insure our officers and directors, we may not be able to retain or recruit qualified officers and directors to manage us.
| 13 |
Risks Related to Our Intellectual Property
The termination of the National Institute of Health, or NIH, license agreement between us and NIH due to patent expiration may diminish our proprietary position.
As a result of the termination of the NIH license agreement between us and the NIH in June 2015 due to patent expiration, we no longer hold rights to a family of composition of matter patents relating to Piclidenoson hat were licensed from NIH. Nevertheless, because Piclidenoson may be a new chemical entity, or NCE, following approval of an NDA, we, if we are the first applicant to obtain NDA approval, may be entitled to five years of data exclusivity in the United States with respect to such NCEs. Analogous data and market exclusivity provisions, of varying duration, may be available in Europe and other foreign jurisdictions. We also have rights under our pharmaceutical use issued patents with respect to Piclidenoson and Namodenoson, which provide patent exclusivity within our field of activity until the mid- to late-2020s. While we believe that we may be able to protect our exclusivity through such use patent portfolio and such period of exclusivity, the lack of composition of matter patent protection may diminish our ability to maintain a proprietary position for our intended uses of Piclidenoson. Moreover, we cannot be certain that we will be the first applicant to obtain an FDA approval for any indication of Piclidenoson and we cannot be certain that we will be entitled to NCE exclusivity. In addition, we have discontinued the prosecution of a family of pending patent applications under joint ownership of us and NIH pertaining to the use of A3AR agonists for the treatment of uveitis. Such diminution of our proprietary position could have a material adverse effect on our business, results of operation and financial condition.
We license from Leiden University intellectual property which protects certain small molecules which target the A3AR, in furtherance of our platform technology, and we could lose our rights to this license if a dispute with Leiden University arises or if we fail to comply with the financial and other terms of the license.
We have licensed intellectual property from Leiden University pursuant to a license agreement. The license agreement imposes certain payment, reporting, confidentiality and other obligations on us. In the event that we were to breach any of the obligations and fail to cure, Leiden University would have the right to terminate the license agreement. In addition, Leiden University has the right to terminate the license agreement upon our bankruptcy, insolvency, or receivership. If any dispute arises with respect to our arrangements with Leiden University, such dispute may disrupt our operations and may have a material adverse impact on us if resolved in a manner that is unfavorable to us.
The failure to obtain or maintain patents, licensing agreements, including our current licensing agreements, and other intellectual property could impact our ability to compete effectively.
To compete effectively, we need to develop and maintain a proprietary position with regard to our own technologies, intellectual property, licensing agreements, product candidates and business. Legal standards relating to the validity and scope of claims in the biotechnology and biopharmaceutical fields are still evolving. Therefore, the degree of future protection for our proprietary rights in our core technologies and any products that might be made using these technologies is also uncertain. The risks and uncertainties that we face with respect to our patents and other proprietary rights include the following:
| ● | while the patents we license have been issued, the pending patent applications we have filed may not result in issued patents or may take longer than we expect to result in issued patents; |
| ● | we may be subject to interference proceedings; |
| ● | we may be subject to opposition proceedings in foreign countries; |
| ● | any patents that are issued may not provide meaningful protection; |
| ● | we may not be able to develop additional proprietary technologies that are patentable; |
| ● | other companies may challenge patents licensed or issued to us or our customers; |
| ● | other companies may independently develop similar or alternative technologies, or duplicate our technologies; |
| ● | other companies may design around technologies we have licensed or developed; and |
| ● | enforcement of patents is complex, uncertain and expensive. |
If patent rights covering our products and methods are not sufficiently broad, they may not provide us with any protection against competitors with similar products and technologies. Furthermore, if the United States Patent and Trademark Office, or the USPTO, or foreign patent officers issue patents to us or our licensors, others may challenge the patents or design around the patents, or the patent office or the courts may invalidate the patents. Thus, any patents we own or license from or to third parties may not provide any protection against our competitors.
| 14 |
We cannot be certain that patents will be issued as a result of any pending applications, and we cannot be certain that any of our issued patents will give us adequate protection from competing products. For example, issued patents, including the patents licensed by us, may be circumvented or challenged, declared invalid or unenforceable, or narrowed in scope. In addition, since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain that we were the first to make our inventions or to file patent applications covering those inventions.
It is also possible that others may obtain issued patents that could prevent us from commercializing our products or require us to obtain licenses requiring the payment of significant fees or royalties in order to enable us to conduct our business. As to those patents that we have licensed, our rights depend on maintaining our obligations to the licensor under the applicable license agreement, and we may be unable to do so.
In addition to patents and patent applications, we depend upon trade secrets and proprietary know-how to protect our proprietary technology. We require our employees, consultants, advisors and collaborators to enter into confidentiality agreements that prohibit the disclosure of confidential information to any other parties. We require our employees and consultants to disclose and assign to us their ideas, developments, discoveries and inventions. These agreements may not, however, provide adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure.
Costly litigation may be necessary to protect our intellectual property rights and we may be subject to claims alleging the violation of the intellectual property rights of others.
We may face significant expense and liability as a result of litigation or other proceedings relating to patents and other intellectual property rights of others. In the event that another party has also filed a patent application or been issued a patent relating to an invention or technology claimed by us in pending applications, we may be required to participate in an interference proceeding declared by the USPTO to determine priority of invention, which could result in substantial uncertainties and costs for us, even if the eventual outcome were favorable to us. We, or our licensors, also could be required to participate in interference proceedings involving issued patents and pending applications of another entity. An adverse outcome in an interference proceeding could require us to cease using the technology or to license rights from prevailing third parties.
The cost to us of any patent litigation or other proceeding relating to our licensed patents or patent applications, even if resolved in our favor, could be substantial. Our ability to enforce our patent protection could be limited by our financial resources, and may be subject to lengthy delays. If we are unable to effectively enforce our proprietary rights, or if we are found to infringe the rights of others, we may be in breach of our License Agreement.
A third party may claim that we are using inventions claimed by their patents and may go to court to stop us from engaging in our normal operations and activities, such as research, development and the sale of any future products. Such lawsuits are expensive and would consume time and other resources. There is a risk that the court will decide that we are infringing the third party’s patents and will order us to stop the activities claimed by the patents, redesign our products or processes to avoid infringement or obtain licenses (which may not be available on commercially reasonable terms). In addition, there is a risk that a court will order us to pay the other party damages for having infringed their patents.
Moreover, there is no guarantee that any prevailing patent owner would offer us a license so that we could continue to engage in activities claimed by the patent, or that such a license, if made available to us, could be acquired on commercially acceptable terms. In addition, third parties may, in the future, assert other intellectual property infringement claims against us with respect to our product candidates, technologies or other matters.
| 15 |
We rely on confidentiality agreements that could be breached and may be difficult to enforce, which could result in third parties using our intellectual property to compete against us.
Although we believe that we take reasonable steps to protect our intellectual property, including the use of agreements relating to the non-disclosure of confidential information to third parties, as well as agreements that purport to require the disclosure and assignment to us of the rights to the ideas, developments, discoveries and inventions of our employees and consultants while we employ them, the agreements can be difficult and costly to enforce. Although we seek to obtain these types of agreements from our contractors, consultants, advisors and research collaborators, to the extent that employees and consultants utilize or independently develop intellectual property in connection with any of our projects, disputes may arise as to the intellectual property rights associated with our products. If a dispute arises, a court may determine that the right belongs to a third party. In addition, enforcement of our rights can be costly and unpredictable. We also rely on trade secrets and proprietary know-how that we seek to protect in part by confidentiality agreements with our employees, contractors, consultants, advisors or others. Despite the protective measures we employ, we still face the risk that:
| ● | these agreements may be breached; |
| ● | these agreements may not provide adequate remedies for the applicable type of breach; |
| ● | our trade secrets or proprietary know-how will otherwise become known; or |
| ● | our competitors will independently develop similar technology or proprietary information. |
International patent protection is particularly uncertain, and if we are involved in opposition proceedings in foreign countries, we may have to expend substantial sums and management resources.
Patent law outside the United States is different than in the United States. Further, the laws of some foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States, if at all. A failure to obtain sufficient intellectual property protection in any foreign country could materially and adversely affect our business, results of operations and future prospects. Moreover, we may participate in opposition proceedings to determine the validity of our foreign patents or our competitors’ foreign patents, which could result in substantial costs and divert management’s resources and attention.
Although most jurisdictions in which we have applied for, intend to apply for, or have been issued patents have patent protection laws similar to those of the United States, some of them do not. For example, we expect to do business in Brazil and India in the future. However, the Brazilian drug regulatory agency, ENVISA, has the authority to nullify patents on the basis of its perceived public interest and the Indian patent law does not allow patent protection for new uses of pharmaceuticals (many of our current patent applications are of such nature). Additionally, due to uncertainty in patent protection law, we have not filed applications in many countries where significant markets exist, including Indonesia, Pakistan, Russia, African countries and Taiwan.
We may be unable to protect the intellectual property rights of the third parties from whom we license certain of our intellectual property or with whom we have entered into other strategic relationships.
Certain of our intellectual property rights are currently licensed from Leiden University, and, in the future, we intend to continue to license intellectual property from Leiden University and/or other universities and/or strategic partners. Such third parties may determine not to protect the intellectual property rights that we license from them and we may be unable defend such intellectual property rights on our own or we may have to undertake costly litigation to defend the intellectual property rights of such third parties. There can be no assurances that we will continue to have proprietary rights to any of the intellectual property that we license from such third parties or otherwise have the right to use through similar strategic relationships. Any loss or limitations on use with respect to our right to use such intellectual property licensed from third parties or otherwise obtained from third parties with whom we have entered into strategic relationships could have a material adverse effect on our business, results of operations and financial condition.
Under current U.S. and Israeli law, we may not be able to enforce employees’ covenants not to compete and therefore may be unable to prevent our competitors from benefiting from the expertise of some of our former employees.
We have entered into non-competition agreements with our key employees, in most cases within the framework of their employment agreements. These agreements prohibit our key employees, if they cease working for us, from competing directly with us or working for our competitors for a limited period. Under applicable U.S. and Israeli law, we may be unable to enforce these agreements. If we cannot enforce our non-competition agreements with our employees, then we may be unable to prevent our competitors from benefiting from the expertise of our former employees, which could materially adversely affect our business, results of operations and ability to capitalize on our proprietary information.
| 16 |
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
| ● | Others may be able to make compounds that are the same as or similar to our product candidates but that are not covered by the claims of the patents that we own or have exclusively licensed; |
| ● | We or our licensors or any future strategic partners might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or have exclusively licensed; |
| ● | We or our licensors or any future strategic partners might not have been the first to file patent applications covering certain of our inventions; |
| ● | Others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| ● | It is possible that our pending patent applications will not lead to issued patents; |
| ● | Issued patents that we own or have exclusively licensed may not provide us with any competitive advantages, or may be held invalid or unenforceable, as a result of legal challenges by our competitors; |
| ● | Our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| ● | We may not develop additional proprietary technologies that are patentable; and |
| ● | The patents of others may have an adverse effect on our business. |
| 17 |
We may be subject to claims challenging the inventorship of our patents and other intellectual property.
We may be subject to claims that former employees, collaborators or other third parties have an interest in our patents or other intellectual property as an inventor or co-inventor. For example, we may have inventorship disputes arise from conflicting obligations of consultants or others who are involved in developing our product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Risks Related to Our Industry
We are subject to government regulations and we may experience delays in obtaining required regulatory approvals in the United States to market our proposed product candidates.
Various aspects of our operations are subject to federal, state or local laws, rules and regulations, any of which may change from time to time. Costs arising out of any regulatory developments could be time-consuming and expensive and could divert management resources and attention and, consequently, could adversely affect our business operations and financial performance.
Delays in regulatory approval, limitations in regulatory approval and withdrawals of regulatory approval may have a material adverse effect on us. If we experience significant delays in testing or receiving approvals or sign-offs to conduct clinical trials, our product development costs, or our ability to license product candidates, will increase. If the FDA grants regulatory approval to market a product, this approval will be limited to those disease states and conditions for which the product has demonstrated, through clinical trials, to be safe and effective. Any product approvals that we receive in the future could also include significant restrictions on the use or marketing of our products. Product approvals, if granted, can be withdrawn for failure to comply with regulatory requirements or upon the occurrence of adverse events following commercial introduction of the products. Failure to comply with applicable FDA or other applicable regulatory requirements may result in criminal prosecution, civil penalties, recall or seizure of products, total or partial suspension of production or injunction, as well as other regulatory action against our product candidates or us. If approval is withdrawn for a product, or if a product were seized or recalled, we would be unable to sell or license that product and our revenues would suffer. In addition, outside the United States, our ability to market any of our potential products is contingent upon receiving market application authorizations from the appropriate regulatory authorities and these foreign regulatory approval processes include all of the risks associated with the FDA approval process described above.
We expect the healthcare industry to face increased limitations on reimbursement as a result of healthcare reform, which could adversely affect third-party coverage of our products and how much or under what circumstances healthcare providers will prescribe or administer our products.
In both the United States and other countries, sales of our products will depend in part upon the availability of reimbursement from third-party payors, which include governmental authorities, managed care organizations and other private health insurers. Third-party payors are increasingly challenging the price and examining the cost effectiveness of medical products and services.
Increasing expenditures for healthcare have been the subject of considerable public attention in the United States. Both private and government entities are seeking ways to reduce or contain healthcare costs. Numerous proposals that would effect changes in the U.S. healthcare system have been introduced or proposed in Congress and in some state legislatures, including reducing reimbursement for prescription products and reducing the levels at which consumers and healthcare providers are reimbursed for purchases of pharmaceutical products.
| 18 |
In 2010, the United States Congress enacted the Patient Protection and Affordable Care Act of 2010 or, Affordable Care Act. The Affordable Care Act seeks to reduce the federal deficit and the rate of growth in health care spending through, among other things, stronger prevention and wellness measures, increased access to primary care, changes in health care delivery systems and the creation of health insurance exchanges. Enrollment in the health insurance exchanges began in October 2013. The Affordable Care Act requires the pharmaceutical industry to share in the costs of reform, by, among other things, increasing Medicaid rebates and expanding Medicaid rebates to cover Medicaid managed care programs. Other components of healthcare reform include funding of pharmaceutical costs for Medicare patients in excess of the prescription drug coverage limit and below the catastrophic coverage threshold. Under the Affordable Care Act, pharmaceutical companies are now obligated to fund 50% of the patient obligation for branded prescription pharmaceuticals in this gap, or “donut hole.” Legislative changes to the Affordable Care Act also remain possible and appear likely in the 115th U.S. Congress under the Trump Administration. Additionally, commencing in 2011, an excise tax was levied against certain branded pharmaceutical products. The tax is specified by statute to be approximately $3 billion in 2012 through 2016, $3.5 billion in 2017, $4.2 billion in 2018, and $2.8 billion each year thereafter. The tax is to be apportioned to qualifying pharmaceutical companies based on an allocation of their governmental programs as a portion of total pharmaceutical government programs.
Although we cannot predict the full effect on our business of the implementation of existing legislation, including the Affordable Care Act or the enactment of additional legislation, we believe that legislation or regulations that reduce reimbursement for or restrict coverage of our products could adversely affect how much or under what circumstances healthcare providers will prescribe or administer our products. This could materially and adversely affect our business by reducing our ability to generate revenue, raise capital, obtain additional collaborators and market our products, following marketing approval. In addition, we believe the increasing emphasis on managed care in the United States has and will continue to put pressure on the price and usage of pharmaceutical products, which may adversely impact any future product sales.
We are subject to federal anti-kickback laws and regulations. Our failure to comply with these laws and regulations could have adverse consequences to us.
There are extensive U.S. federal and state laws and regulations prohibiting fraud and abuse in the healthcare industry that can result in significant criminal and civil penalties. These federal laws include: the anti-kickback statute, which prohibits certain business practices and relationships, including the payment or receipt of remuneration for the referral of patients whose care will be paid by Medicare or other federal healthcare programs; the physician self-referral prohibition, commonly referred to as the Stark Law; the anti-inducement law, which prohibits providers from offering anything to a Medicare or Medicaid beneficiary to induce that beneficiary to use items or services covered by either program; the False Claims Act, which prohibits any person from knowingly presenting or causing to be presented false or fraudulent claims for payment by the federal government, including the Medicare and Medicaid programs; and the Civil Monetary Penalties Law, which authorizes the U.S. Department of Health and Human Services to impose civil penalties administratively for fraudulent or abusive acts.
Sanctions for violating these federal laws include criminal and civil penalties that range from punitive sanctions, damage assessments, money penalties, imprisonment, denial of Medicare and Medicaid payments, or exclusion from the Medicare and Medicaid programs, or both, and debarment. As federal and state budget pressures continue, federal and state administrative agencies may also continue to escalate investigation and enforcement efforts to root out waste and to control fraud and abuse in governmental healthcare programs. Private enforcement of healthcare fraud has also increased, due in large part to amendments to the civil False Claims Act in 1986 that were designed to encourage private persons to sue on behalf of the government. A violation of any of these federal and state fraud and abuse laws and regulations could have a material adverse effect on our liquidity and financial condition. An investigation into the use by physicians of any of our products once commercialized may dissuade physicians from either purchasing or using them, and could have a material adverse effect on our ability to commercialize those products.
| 19 |
Risks Related to our Ordinary Shares and ADSs
We may be a passive foreign investment company, or PFIC, for U.S. federal income tax purposes in 2016 or in any subsequent year. There may be negative tax consequences for U.S. taxpayers that are holders of our ordinary shares or the ADSs.
We will be treated as a PFIC for U.S. federal income tax purposes in any taxable year in which either (i) at least 75% of our gross income is “passive income” or (ii) on average at least 50% of our assets by value produce passive income or are held for the production of passive income. Passive income for this purpose generally includes, among other things, certain dividends, interest, royalties, rents and gains from commodities and securities transactions and from the sale or exchange of property that gives rise to passive income. Passive income also includes amounts derived by reason of the temporary investment of funds, including those raised in a public offering. In determining whether a non-U.S. corporation is a PFIC, a proportionate share of the income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account. We believe we may be a PFIC during 2016 and although we have not determined whether we will be a PFIC in 2017, or in any subsequent year, our operating results for any such years may cause us to be a PFIC. If we are a PFIC in 2016, or any subsequent year, and a U.S. shareholder does not make an election to treat us as a “qualified electing fund,” or QEF, or make a “mark-to-market” election, then “excess distributions” to a U.S. shareholder, and any gain realized on the sale or other disposition of our ordinary shares or ADSs will be subject to special rules. Under these rules: (i) the excess distribution or gain would be allocated ratably over the U.S. shareholder’s holding period for the ordinary shares (or ADSs, as the case may be); (ii) the amount allocated to the current taxable year and any period prior to the first day of the first taxable year in which we were a PFIC would be taxed as ordinary income; and (iii) the amount allocated to each of the other taxable years would be subject to tax at the highest rate of tax in effect for the applicable class of taxpayer for that year, and an interest charge for the deemed deferral benefit would be imposed with respect to the resulting tax attributable to each such other taxable year. In addition, if the U.S. Internal Revenue Service determines that we are a PFIC for a year with respect to which we have determined that we were not a PFIC, it may be too late for a U.S. shareholder to make a timely QEF or mark-to-market election. U.S. shareholders who hold our ordinary shares or ADSs during a period when we are a PFIC will be subject to the foregoing rules, even if we cease to be a PFIC in subsequent years, subject to exceptions for U.S. shareholders who made a timely QEF or mark-to-market election. A U.S. shareholder can make a QEF election by completing the relevant portions of and filing IRS Form 8621 in accordance with the instructions thereto. Upon request, we will annually furnish U.S. shareholders with information needed in order to complete IRS Form 8621 (which form would be required to be filed with the IRS on an annual basis by the U.S. shareholder) and to make and maintain a valid QEF election for any year in which we or any of our subsidiaries that we control is a PFIC.
Issuance of additional equity securities may adversely affect the market price of the ADSs or ordinary shares.
We are currently authorized to issue 80,000,000 ordinary shares. As of March 29, 2017, we had 32,709,901 ordinary shares issued and outstanding, excluding treasury shares and we had no preferred shares outstanding. As of March 29, 2017, we also had warrants to purchase 13,608,824 ordinary shares and options to purchase 737,028 ordinary shares outstanding, of which options to purchase 462,499 ordinary shares are currently fully vested or vest within the next 60 days.
To the extent that ADSs or ordinary shares are issued or options and warrants are exercised, holders of the ADSs and our ordinary shares will experience dilution. In addition, in the event of any future issuances of equity securities or securities convertible into or exchangeable for ADSs or ordinary shares, holders of the ADSs or our ordinary shares may experience dilution. We also consider from time to time various strategic alternatives that could involve issuances of additional ADSs or ordinary shares, including but not limited to acquisitions and business combinations, but do not currently have any definitive plans to enter into any of these transactions.
We have no plans to pay dividends on our ordinary shares, and you may not receive funds without selling the ADSs or ordinary shares.
We have not declared or paid any cash dividends on our ordinary shares, nor do we expect to pay any cash dividends on our ordinary shares for the foreseeable future. We currently intend to retain any additional future earnings to finance our operations and growth and for future stock repurchases and, therefore, we have no plans to pay cash dividends on our ordinary shares at this time. Any future determination to pay cash dividends on our ordinary shares will be at the discretion of our board of directors and will be dependent on our earnings, financial condition, operating results, capital requirements, any contractual restrictions, and other factors that our board of directors deems relevant. Accordingly, you may have to sell some or all of the ADSs or ordinary shares in order to generate cash from your investment. You may not receive a gain on your investment when you sell the ADSs or ordinary shares and may lose the entire amount of your investment.
The market price of our ordinary shares and ADSs is subject to fluctuation, which could result in substantial losses by our investors.
The stock market in general and the market price of our ordinary shares on the TASE and our ADSs on the NYSE MKT is subject to fluctuation, and changes in our share price may be unrelated to our operating performance. The market price of our ordinary shares and ADSs are and will be subject to a number of factors, including:
| ● | announcements of technological innovations or new products by us or others; | |
| ● | announcements by us of significant strategic partnerships, out-licensing, in-licensing, joint ventures, acquisitions or capital commitments; | |
|
● | expiration or terminations of licenses, research contracts or other collaboration agreements; |
| 20 |
| ● | public concern as to the safety of drugs we, our licensees or others develop; | |
| ● | general market conditions; | |
| ● | the volatility of market prices for shares of biotechnology companies generally; | |
| ● | success of research and development projects; | |
| ● | success in clinical and preclinical studies; | |
| ● | departure of key personnel; |
| ● | developments concerning intellectual property rights or regulatory approvals; | |
| ● | variations in our and our competitors’ results of operations; | |
| ● | changes in earnings estimates or recommendations by securities analysts, if our ordinary shares or ADSs are covered by analysts; | |
| ● | changes in government regulations or patent decisions; | |
●
● |
developments by our licensees; and
general market conditions and other factors, including factors unrelated to our operating performance. |
These factors and any corresponding price fluctuations may materially and adversely affect the market price of our ordinary shares and the ADSs and result in substantial losses by our investors.
Additionally, market prices for securities of biotechnology and pharmaceutical companies historically have been very volatile. The market for these securities has from time to time experienced significant price and volume fluctuations for reasons unrelated to the operating performance of any one company. In the past, following periods of market volatility, shareholders have often instituted securities class action litigation. If we were involved in securities litigation, it could have a substantial cost and divert resources and attention of management from our business, even if we are successful. Future sales of our ordinary shares or ADSs could reduce the market price of our ordinary shares and ADSs.
Substantial sales of our ordinary shares or the ADSs either on the TASE or on the NYSE MKT, as applicable, may cause the market price of our ordinary shares or the ADSs to decline.
Sales by us or our security-holders of substantial amounts of our ordinary shares or the ADSs, or the perception that these sales may occur in the future, could cause a reduction in the market price of our ordinary shares or the ADSs. The issuance of any additional ordinary shares or ADSs, or any securities that are exercisable for or convertible into our ordinary shares or the ADSs, may have an adverse effect on the market price of our ordinary shares or the ADSs, as applicable, and will have a dilutive effect on our shareholders.
ADS holders are not shareholders and do not have shareholder rights.
The Bank of New York Mellon, as Depositary, delivers the ADSs. Each ADS represents two of our ordinary shares. ADS holders will not be treated as shareholders and do not have the rights of shareholders. The depositary will be the holder of the shares underlying the ADSs. Holders of ADSs will have ADS holder rights. A deposit agreement among us, the depositary, ADS holders and the beneficial owners of ADSs sets out ADS holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and the ADSs. Our shareholders have shareholder rights. Israeli law and our Articles of Association govern shareholder rights. ADS holders do not have the same voting rights as our shareholders. Shareholders are entitled to our notices of general meetings and to attend and vote at our general meetings of shareholders. At a general meeting, every shareholder present (in person or by proxy, attorney or representative) and entitled to vote has one vote. This is subject to any other rights or restrictions which may be attached to any shares. ADS holders may instruct the depositary how to vote the number of deposited shares their ADSs represent. Otherwise you won’t be able to exercise your right to vote unless you withdraw the shares. However, you may not know about the meeting enough in advance to withdraw the shares. The depositary will notify ADS holders of shareholders’ meetings and arrange to deliver our voting materials to them if we ask it to. Those materials will describe the matters to be voted on and explain how ADS holders may instruct the depositary how to vote. For instructions to be valid, they must reach the depositary by a date set by the depositary. The depositary will try, as far as practical, subject to the laws of Israel and our articles of association or similar documents, to vote or to have its agents vote the shares or other deposited securities as instructed by ADS holders. The depositary will only vote or attempt to vote as instructed. We cannot assure you that you will receive the voting materials in time to ensure that you can instruct the depositary to vote your hares. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the matter of carrying out voting instructions. This means that you may not be able to exercise your right to vote and there may be nothing you can do if your shares are not voted as requested.
| 21 |
ADS holders do not have the same rights to receive dividends or other distributions as our shareholders. Subject to any special rights or restrictions attached to a share, the directors may determine that a dividend will be payable on a share and fix the amount, the time for payment and the method for payment (although we have never declared or paid any cash dividends on our ordinary shares and we do not anticipate paying any cash dividends in the foreseeable future). Dividends and other distributions payable to our shareholders with respect to our ordinary shares generally will be payable directly to them. Any dividends or distributions payable with respect to ordinary shares deposited in the ADS facility will be paid to the depositary, which has agreed to pay to ADS holders the cash dividends or other distributions it or the custodian receives on shares or other deposited securities, after deducting its fees and expenses. ADS holders will receive these distributions in proportion to the number of ordinary shares their ADSs represent. In addition, there may be certain circumstances in which the depositary may not pay ADS holders amounts distributed by us as a dividend or distribution.
Our ordinary shares and the ADSs are traded on different markets and this may result in price variations.
Our ordinary shares have traded on the TASE since October 2005 and the ADSs have been listed on the NYSE MKT since November 2013. Trading on these markets will take place in different currencies (U.S. dollars on the NYSE MKT and NIS on the TASE), and at different times (resulting from different time zones, different trading days and different public holidays in the United States and Israel). The trading prices of our securities on these two markets may differ due to these and other factors. Any decrease in the price of our securities on one of these markets could cause a decrease in the trading price of our securities on the other market.
The ADSs have a limited prior trading history in the United States, and an active market may not develop, which may limit the ability of our investors to sell the ADSs in the United States.
There is a limited public market for the ADSs in the United States. The ADSs are thinly traded and an active trading market for the ADSs may never develop or may not be sustained if one develops. If an active market for the ADSs does not develop or is not sustained, it may be difficult to sell your ADSs.
We have incurred significant additional increased costs as a result of the listing of ADSs for trading on the NYSE MKT, and our management is required to devote substantial time to new compliance initiatives as well as to compliance with ongoing U.S. and Israeli reporting requirements.
As a public company in the United States, we incur additional significant accounting, legal and other expenses that we did not incur before becoming a reporting company in the United States. We also incur costs associated with corporate governance requirements of the SEC and the NYSE MKT Company Guide, as well as requirements under Section 404 and other provisions of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act as a result of ADSs being listed on the NYSE MKT. These rules and regulations have increased our legal and financial compliance costs, introduced new costs such as investor relations, stock exchange listing fees and shareholder reporting, and made some activities more time consuming and costly. The implementation and testing of such processes and systems may require us to hire outside consultants and incur other significant costs. Any future changes in the laws and regulations affecting public companies in the United States and Israel, including Section 404 and other provisions of the Sarbanes-Oxley Act, the rules and regulations adopted by the SEC and the NYSE MKT Company Guide, as well as applicable Israeli reporting requirements, for so long as they apply to us, may result in increased costs to us as we respond to such changes. These laws, rules and regulations could make it more difficult or more costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our Board of Directors, our board committees or as executive officers.
| 22 |
As a foreign private issuer, we are permitted to follow certain home country corporate governance practices instead of applicable SEC and NYSE MKT requirements, which may result in less protection than is accorded to investors under rules applicable to domestic issuers.
As a foreign private issuer, we will be permitted to follow certain home country corporate governance practices instead of those otherwise required under the NYSE MKT Company Guide for domestic issuers. For instance, we may follow home country practice in Israel with regard to, among other things, composition and function of the audit committee and other committees of our Board of Directors and certain general corporate governance matters. In addition, in certain instances we will follow our home country law, instead of the NYSE MKT Company Guide, which requires that we obtain shareholder approval for certain dilutive events, such as an issuance that will result in a change of control of the company, certain transactions other than a public offering involving issuances of a 20% or more interest in the company and certain acquisitions of the stock or assets of another company. We comply with the director independence requirements of the NYSE MKT Company Guide, including the requirement that a majority of the Board of Directors be independent, and make the required affirmative determination thereunder upon filing the listing application with the NYSE MKT. Following our home country governance practices as opposed to the requirements that would otherwise apply to a United States company listed on the NYSE MKT may provide less protection than is accorded to investors under the NYSE MKT Company Guide applicable to domestic issuers.
In addition, as a foreign private issuer, we will be exempt from the rules and regulations under the U.S. Securities Exchange Act of 1934, as amended, or the Exchange Act, related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders will be exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we will not be required under the Exchange Act to file annual, quarterly and current reports and financial statements with the SEC as frequently or as promptly as domestic companies whose securities are registered under the Exchange Act.
Because we became a reporting company under the Exchange Act by means of filing a Form 20-F, we may have difficulty attract the attention of research analysts at major brokerage firms.
Because we did not become a reporting company by conducting an underwritten initial public offering in the U.S., we may have difficulty attracting the attention of security analysts at major brokerage firms in order for them to provide coverage of our company. The failure to receive research coverage or support in the market for our shares will have an adverse effect on our ability to develop a liquid market for the ADSs.
If we are unable to satisfy the requirements of Section 404 of the Sarbanes-Oxley Act as they apply to a foreign private issuer that is listed on a U.S. exchange, or our internal control over financial reporting is not effective, the reliability of our financial statements may be questioned and our share price and the ADS price may suffer.
We have become subject to the requirements of the Sarbanes-Oxley Act since the ADSs are listed on the NYSE MKT. Section 404 of the Sarbanes-Oxley Act requires companies subject to the reporting requirements of the U.S. securities laws to do a comprehensive evaluation of its and its subsidiaries’ internal control over financial reporting. To comply with this statute, we must document and test our internal control procedures and our management and issue a report concerning our internal control over financial reporting. In addition, under the JOBS Act, emerging growth companies, like ourselves, are exempt from certain reporting requirements, including the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act. Under this exemption, our auditor will not be required to attest to and report on our management’s assessment of our internal control over financial reporting during a five-year transition period. We will need to prepare for compliance with Section 404 by strengthening, assessing and testing our system of internal controls to provide the basis for our report. However, the continuous process of strengthening our internal controls and complying with Section 404 is complicated and time-consuming. Furthermore, as our business continues to grow both domestically and internationally, our internal controls will become more complex and will require significantly more resources and attention to ensure our internal controls remain effective overall. During the course of the testing, our management may identify material weaknesses or significant deficiencies, which may not be remedied in a timely manner to meet the deadline imposed by the Sarbanes-Oxley Act. If our management cannot favorably assess the effectiveness of our internal control over financial reporting, or our independent registered public accounting firm identifies material weaknesses in our internal controls, investor confidence in our financial results may weaken, and the market price of our securities may suffer.
| 23 |
As an “emerging growth company” under the JOBS Act, we are permitted to, and intend to, rely on exemptions from certain disclosure requirements.
As an “emerging growth company” under the JOBS Act, we are permitted to, and intend to, rely on exemptions from certain disclosure requirements. We are an emerging growth company until the earliest of: (i) the last day of the fiscal year during which we had total annual gross revenues of $1 billion or more, (ii) the last day of the fiscal year following the fifth anniversary of the date of the first sale of our common stock pursuant to an effective registration statement, (iii) the date on which we have, during the previous three-year period, issued more than $1 billion in non-convertible debt or (iv) the date on which we are deemed a “large accelerated issuer” as defined in Regulation S-K of the Securities Act. For so long as we remain an emerging growth company, we will not be required to:
| ● | have an auditor report on our internal control over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act; |
| ● | comply with any requirement that may be adopted by the Public Company Accounting Oversight Board, or the PCAOB, regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements (auditor discussion and analysis); |
| ● | submit certain executive compensation matters to shareholders advisory votes pursuant to the “say on frequency” and “say on pay” provisions (requiring a non-binding shareholder vote to approve compensation of certain executive officers) and the “say on golden parachute” provisions (requiring a non-binding shareholder vote to approve golden parachute arrangements for certain executive officers in connection with mergers and certain other business combinations) of the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010; and |
| ● | include detailed compensation discussion and analysis in our filings under the Exchange Act, and instead may provide a reduced level of disclosure concerning executive compensation. |
Although we intend to rely on the exemptions provided in the JOBS Act, the exact implications of the JOBS Act for us are still subject to interpretations and guidance by the SEC and other regulatory agencies. In addition, as our business grows, we may no longer satisfy the conditions of an emerging growth company. We are currently evaluating and monitoring developments with respect to these new rules and we cannot assure you that we will be able to take advantage of all of the benefits from the JOBS Act.
Risks Related to our Operations in Israel
We conduct our operations in Israel and therefore our results may be adversely affected by political, economic and military instability in Israel and its region.
Our headquarters, all of our operations and some of our suppliers and third party contractors are located in central Israel and our key employees, officers and most of our directors are residents of Israel. Accordingly, political, economic and military conditions in Israel and the surrounding region may directly affect our business. Since the establishment of the State of Israel in 1948, a number of armed conflicts have taken place between Israel and its Arab neighbors. Any hostilities involving Israel or the interruption or curtailment of trade within Israel or between Israel and its trading partners could adversely affect our operations and results of operations and could make it more difficult for us to raise capital. During the winter of 2008, winter of 2012 and the summer of 2014, Israel was engaged in an armed conflict with Hamas, a militia group and political party operating in the Gaza Strip, and during the summer of 2006, Israel was engaged in an armed conflict with Hezbollah, a Lebanese Islamist Shiite militia group and political party. Israel faces political tension with respect to its relationships with Turkey, Iran and certain Arab neighbor countries. In addition, recent conflicts involved missile strikes against civilian targets in various parts of Israel, and negatively affected business conditions in Israel. Recent political uprisings and social unrest in various countries in the Middle East and North Africa are affecting the political stability of those countries. This instability may lead to deterioration of the political relationships that exist between Israel and these countries, and have raised concerns regarding security in the region and the potential for armed conflict. Any armed conflicts, terrorist activities or political instability in the region could adversely affect business conditions and could harm our results of operations. For example, any major escalation in hostilities in the region could result in a portion of our employees and service providers being called up to perform military duty for an extended period of time. Parties with whom we do business have sometimes declined to travel to Israel during periods of heightened unrest or tension, forcing us to make alternative arrangements when necessary. In addition, the political and security situation in Israel may result in parties with whom we have agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions in such agreements. Any future deterioration in the political and security situation in Israel will negatively impact our business.
| 24 |
Our commercial insurance does not cover losses that may occur as a result of events associated with the security situation in the Middle East. Although the Israeli government currently covers the reinstatement value of direct damages that are caused by terrorist attacks or acts of war, we cannot assure you that this government coverage will be maintained. Any losses or damages incurred by us could have a material adverse effect on our business. Any armed conflicts or political instability in the region would likely negatively affect business conditions and could harm our results of operations.
Further, in the past, the State of Israel and Israeli companies have been subjected to an economic boycott. Several countries still restrict business with the State of Israel and with Israeli companies. These restrictive laws and policies may have an adverse impact on our operating results, financial condition or the expansion of our business.
Our operations may be disrupted as a result of the obligation of Israeli citizens to perform military service.
Many Israeli citizens, including Motti Farbstein, our Chief Operating and Financial Officer, are obligated to perform one month, and in some cases more, of annual military reserve duty until they reach the age of 45 (or older, for reservists with certain occupations) and, in the event of a military conflict, may be called to active duty. In response to increases in terrorist activity, there have been periods of significant call-ups of military reservists. It is possible that there will be military reserve duty call-ups in the future. Our operations could be disrupted by such call-ups, which may include the call-up of Motti Farbstein. Such disruption could materially adversely affect our business, financial condition and results of operations.
Because a certain portion of our expenses is incurred in currencies other than the NIS, our results of operations may be harmed by currency fluctuations and inflation.
Our reporting and functional currency is the NIS, and we pay a substantial portion of our expenses in NIS. Part of our expenses are payable in U.S. dollars or in Euros as well as the revenues from our licensing arrangements that are payable in U.S. dollars and Canadian dollars, we expect our revenues from future licensing arrangements to be denominated in U.S. dollars or in Euros. As a result, we are exposed to the currency fluctuation risks relating to the recording of our revenues in NIS. For example, if the NIS strengthens against either the U.S. dollar or the Euro, our reported revenues in NIS may be lower than anticipated. The Israeli rate of inflation has not offset or compounded the effects caused by fluctuations between the NIS and the U.S. dollar or the Euro. To date, we have not engaged in hedging transactions. Although the Israeli rate of inflation has not had a material adverse effect on our financial condition during 2014, 2015, or 2016 to date, we may, in the future, decide to enter into currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rates of the currencies mentioned above in relation to the NIS. These measures, however, may not adequately protect us from material adverse effects.
Provisions of Israeli law may delay, prevent or otherwise impede a merger with, or an acquisition of, our Company, which could prevent a change of control, even when the terms of such a transaction are favorable to us and our shareholders.
Israeli corporate law regulates mergers, requires tender offers for acquisitions of shares above specified thresholds, requires special approvals for transactions involving directors, officers or significant shareholders and regulates other matters that may be relevant to these types of transactions. For example, a merger may not be consummated unless at least 50 days have passed from the date that a merger proposal was filed by each merging company with the Israel Registrar of Companies and at least 30 days from the date that the shareholders of both merging companies approved the merger. In addition, a majority of each class of securities of the target company must approve a merger. Moreover, a full tender offer can only be completed if the acquirer receives at least 95% of the issued share capital; provided that, pursuant to an amendment to the Israeli Companies Law, effective as of May 15, 2011, a majority of the offerees that do not have a personal interest in such tender offer shall have approved the tender offer; except that, if the total votes to reject the tender offer represent less than 2% of our issued and outstanding share capital, in the aggregate, approval by a majority of the offerees that do not have a personal interest in such tender offer is not required to complete the tender offer), and the shareholders, including those who indicated their acceptance of the tender offer, may, at any time within six months following the completion of the tender offer, petition the court to alter the consideration for the acquisition (unless the acquirer stipulated in the tender offer that a shareholder that accepts the offer may not seek appraisal rights).
| 25 |
Furthermore, Israeli tax considerations may make potential transactions unappealing to us or to our shareholders whose country of residence does not have a tax treaty with Israel exempting such shareholders from Israeli tax. For example, Israeli tax law does not recognize tax-free share exchanges to the same extent as U.S. tax law. With respect to mergers, Israeli tax law allows for tax deferral in certain circumstances but makes the deferral contingent on the fulfillment of numerous conditions, including a holding period of two years from the date of the transaction during which sales and dispositions of shares of the participating companies are restricted. Moreover, with respect to certain share swap transactions, the tax deferral is limited in time, and when such time expires, the tax becomes payable even if no actual disposition of the shares has occurred.
These and other similar provisions could delay, prevent or impede an acquisition of us or our merger with another company, even if such an acquisition or merger would be beneficial to us or to our shareholders. See “Item 10. Additional Information — Memorandum and Articles of Association.”
It may be difficult to enforce a U.S. judgment against us and our officers and directors named in this Annual Report on Form 20-F in Israel or the United States, or to serve process on our officers and directors.
We are incorporated in Israel. All of our executive officers and directors listed in this Annual Report on Form 20-F reside outside of the United States, and all of our assets and most of the assets of our executive officers and directors are located outside of the United States. Therefore, a judgment obtained against us or most of our executive officers and all of our directors in the United States, including one based on the civil liability provisions of the U.S. federal securities laws, may not be collectible in the United States and may not be enforced by an Israeli court. It also may be difficult for you to effect service of process on these persons in the United States or to assert U.S. securities law claims in original actions instituted in Israel.
Your rights and responsibilities as a shareholder will be governed by Israeli law which may differ in some respects from the rights and responsibilities of shareholders of U.S. companies.
We are incorporated under Israeli law. The rights and responsibilities of the holders of our ordinary shares and ADSs are governed by our Articles of Association and Israeli law. These rights and responsibilities differ in some respects from the rights and responsibilities of shareholders in typical U.S.-based corporations. In particular, a shareholder of an Israeli company has a duty to act in good faith toward the company and other shareholders and to refrain from abusing its power in the company, including, among other things, in voting at the general meeting of shareholders on matters such as amendments to a company’s articles of association, increases in a company’s authorized share capital, mergers and acquisitions and interested party transactions requiring shareholder approval. In addition, a shareholder who knows that it possesses the power to determine the outcome of a shareholder vote or to appoint or prevent the appointment of a director or executive officer in the company has a duty of fairness toward the company. There is limited case law available to assist us in understanding the implications of these provisions that govern shareholders’ actions. These provisions may be interpreted to impose additional obligations and liabilities on holders of our ordinary shares and ADSs that are not typically imposed on shareholders of U.S. corporations.
ITEM 4. Information on the Company
A. History and Development of the Company
Our legal name is Can-FiteBio Pharma Ltd. and our commercial name is “Can-Fite.” We are a company limited by shares organized under the laws of the State of Israel. Our principal executive offices are located at 10 Bareket Street, Kiryat Matalon, Petah-Tikva 4951778 Israel. Our telephone number is +972 (3) 924-1114.
We were founded on September 11, 1994 by Pnina Fishman, Ph.D., our Chief Executive Officer and a director, and Ilan Cohn, Ph.D., our Chairman of the Board of Directors, under the name Can-Fite Technologies Ltd. On January 7, 2001, we changed our name to Can-FiteBio Pharma Ltd. We completed our initial public offering in Israel in October 2005 and our ordinary shares are traded on the TASE under the symbol “CFBI”. On October 2, 2012, our ADSs began trading over the counter, or OTC, in the United States under the symbol “CANFY” and on November 19, 2013,our ADSs began trading on the NYSE MKT under the symbol “CANF.”
In November 2011, through a series of transactions, we spun-off our activity in the ophthalmic field to OphthaliX, Inc., a Delaware corporation and successor-in-interest to Denali Concrete Management, Inc., a Nevada corporation, or OphthaliX, whose common shares are traded in the United States on OTC under the symbol “OPLI.” In the spin-off transactions, we granted an exclusive license for the use of our Piclidenoson drug candidate in the ophthalmic field, or the License Agreement, to Eye-Fite Ltd., an Israel limited company and a then former wholly-owned subsidiary of ours, or Eye-Fite, and transferred our issued and outstanding ordinary shares in Eye-Fite to OphthaliX in exchange for an 86.7% interest in OphthaliX. In connection with the spin-off transactions, OphthaliX completed a series of private placement financing transactions. Following the spin-off transactions and the private placement financing transactions, we held approximately 82% interest in OphthaliX.
| 26 |
On July 5, 2016, OphthaliX released top-line results from its Phase II trial with respect to the development of Piclidenoson for the treatment of glaucoma or related syndromes of ocular hypertension. In this trial, no statistically significant differences were found between the Piclidenoson treated group and the placebo group in the primary endpoint of lowering intra ocular pressure. Piclidenoson was found to have a favorable safety profile and was well tolerated. Subsequently, in September 2016, OphthaliX’s board of directors and we consented in writing to, among other things, to OphthaliX’s voluntary dissolution and liquidation pursuant to a Plan of Dissolution, that would result in OphthaliX’s complete dissolution and liquidation. The Plan of Dissolution was expected to go into effect 20 days after the date an information statement was first given to all OphthaliX shareholders who did not execute the written consent. Prior to the distribution of the information statement, on November 10, 2016, OphthaliX’s board of directors abandoned its voluntary dissolution and liquidation. On the same day, OphthaliX’s board of directors authorized its entry into a non-binding letter of intent with an Israeli company for the acquisition of such company by way of a reverse triangular merger. Subsequently on November 15, 2016, we entered into the non-binding letter of intent. The proposed reverse merger is subject to signing of definitive transaction documents and the completion of closing conditions. There can be no assurance that the transactions contemplated by the letter of intent will be completed.
OphthaliX has ceased all research and development operations and is considered to be a shell company as defined in Rule 12b-2 of the Securities and Exchange Act of 1934.
Our capital expenditures for the years ended December 31, 2016, 2015 and 2014 were NIS 40,000, NIS 177,000 and NIS 37,000, respectively. Our current capital expenditures are made solely within Israel and primarily consist of the acquisition of computers and related communications equipment. Such capital expenditures are financed internally.
We qualify as an “emerging growth company,” as defined in the JOBS Act. For as long as we are deemed an emerging growth company, we may take advantage of specified reduced reporting and other regulatory requirements that are generally unavailable to other public companies. These provisions include:
| ● | an exemption from the auditor attestation requirement in the assessment of our internal controls over financial reporting required by Section 404 of the Sarbanes-Oxley Act of 2002; |
| ● | an exemption from compliance with any new requirements adopted by the PCAOB requiring mandatory audit firm rotation or a supplement to the auditor’s report in which the auditor would be required to provide additional information about our audit and our financial statements; and |
| ● | reduced disclosure about our executive compensation arrangements. |
We will continue to be deemed an emerging growth company until the earliest of:
| ● | the last day of our fiscal year in which we have total annual gross revenues of $1,000,000,000 (as such amount is indexed for inflation every five years by the SEC to reflect the change in the Consumer Price Index for All Urban Consumers published by the Bureau of Labor Statistics, setting the threshold to the nearest $1,000,000) or more; |
| ● | the last day of our fiscal year following the fifth anniversary of the date of our first sale of common equity securities pursuant to an effective registration statement under the Securities Act; |
| ● | the date on which we have, during the prior three-year period, issued more than $1,000,000,000 in non-convertible debt; or |
| ● | the date on which we are deemed to be a ‘large accelerated filer,” as defined in Regulation S-K under the Securities Act. |
| 27 |
B. Business Overview
We are a clinical-stage biopharmaceutical company focused on developing orally bioavailable small molecule therapeutic products for the treatment of autoimmune inflammatory indications, oncology and liver diseases as well as sexual dysfunction. Our platform technology utilizes the Gi protein associated A3AR as a therapeutic target. A3AR is highly expressed in inflammatory and cancer cells, and not significantly expressed in normal cells, suggesting that the receptor could be a unique target for pharmacological intervention. Our pipeline of drug candidates are synthetic, highly specific agonists and allosteric modulators, or ligands or molecules that initiate molecular events when binding with target proteins, targeting the A3AR.
Our product pipeline is based on the research of Dr. Pnina Fishman, who investigated a clinical observation that tumor metastasis can be found in most body tissues, but are rarely found in muscle tissue, which constitutes approximately 60% of human body weight. Dr. Fishman’s research revealed that one reason that striated muscle tissue is resistant to tumor metastasis is that muscle cells release small molecules which bind with high selectivity to the A3AR. As part of her research, Dr. Fishman also discovered that A3ARs have significant expression in tumor and inflammatory cells, whereas normal cells have low or no expression of this receptor. The A3AR agonists and allosteric modulators, currently our pipeline of drug candidates, bind with high selectivity and affinity to the A3ARs and upon binding to the receptor initiate down-stream signal transduction pathways resulting in apoptosis, or programmed cell death, of tumors and inflammatory cells and to the inhibition of inflammatory cytokines. Cytokines are proteins produced by cells that interact with cells of the immune system in order to regulate the body’s response to disease and infection. Overproduction or inappropriate production of certain cytokines by the body can result in disease.
Our product candidates, CF101, CF102 and CF602 are being developed to treat autoimmune inflammatory indications, oncology and liver diseases as well as sexual dysfunction. CF101, also known as Piclidenoson, is in an advance stage of clinical development for the treatment of autoimmune-inflammatory diseases, including RA and psoriasis. CF102, also known as Namodenoson, is being developed for the treatment of HCC and has orphan drug designation for the treatment of HCC in the U.S. and Europe. Namodenoson was granted Fast Track designation by the FDA as a second line treatment to improve survival for patients with advanced hepatocellular carcinoma who have previously received Nexavar (sorafenib). Namodenoson is also being developed for the treatment of non-alcoholic steatohepatitis, or NASH, following our study which revealed compelling pre-clinical data on Namodenoson in the treatment of NASH, a disease for which no FDA approved therapies currently exist. CF602 is our second generation allosteric drug candidate for the treatment of sexual dysfunction, which has shown efficacy in the treatment of erectile dysfunction in preclinical studies and is being prepared for an IND submission to the FDA and a Phase I trial. Preclinical studies revealed that our drug candidates have potential to treat additional inflammatory diseases, such as Crohn’s disease, oncological diseases and viral diseases, such as the JC virus.
We believe our pipeline of drug candidates represent a significant market opportunity. For instance, according to Visiongain, the world RA market size is predicted to generate revenues of $34.6 billion in 2020 and the psoriasis drug market is forecasted to be worth $8.9 billion by 2018. According to Datamonitor, the HCC drug market is expected to reach $1.4 billion by 2019.
We have in-licensed an allosteric modulator of the A3AR, CF602 from Leiden University. In addition, we have out-licensed Piclidenoson (i) for the treatment of RA to Kwang Dong Pharmaceutical Co. Ltd., a South Korean limited company, or KD for the Korean market, and (ii) for the treatment of psoriasis and RA to Cipher Pharmaceuticals for the Canadian market.
With respect to Namodenoson, in October 2016, we entered into an exclusive distribution agreement with Chong Kun Dang Pharmaceuticals, or CKD for the exclusive right to distribute Namodenoson for the treatment of liver cancer in South Korea, upon receipt of regulatory approvals. The distribution agreement provides for up to $3,000,000 in upfront and milestone payments, plus royalties on net sales of 23%. The distribution agreement further provides that we will deliver finished product to CKD and grant CKD a right of first refusal to distribute Namodenoson for other indications for which we develop Namodenoson. See “Item 4. Information on the Company—Out-Licensing and Distribution Agreements—CKD Agreement”.
In July 2016, OphthaliX released top-line results from its Phase II clinical trial of Piclidenoson for the treatment of glaucoma. In this trial, no statistically significant differences were found between the Piclidenoson treated group and the placebo group in the primary endpoint of lowering intra ocular pressure, or IOP. High IOP is a characteristic of glaucoma. Piclidenoson was found to have a favorable safety profile and was well tolerated. Based on these overall results, OphthaliX sees no immediate path forward in glaucoma. OphthaliX has since ceased all research and development operations.
In June 2015, we received a lawsuit, filed with the District Court of Tel-Aviv, requesting recognition of this lawsuit as a class action. The lawsuit named us, our Chief Executive Officer and directors as defendants. The lawsuit alleges, among other things, that we misled the public with regard to disclosures concerning the efficacy of our drug candidate, Piclidenoson in relation to the Psoriasis studies. The claimant alleges that he suffered personal damages of over NIS 73,000, while also claiming that our shareholders suffered aggregate damages of approximately NIS 125 million. On March 31, 2016, we filed a response to the lawsuit. On March 1, 2017, a hearing was held in the District Court on whether to certify the lawsuit as a class action. A final hearing on the certification is scheduled for April 26, 2017.
| 28 |
We believe that our drug candidates have certain unique characteristics and advantages over drugs currently available on the market and under development to treat these indications. To date, we have generated our pipeline by in-licensing, researching and developing two synthetic A3AR agonists, Piclidenoson and Namodenoson, and an allosteric modulator, CF602. For example, our technology platform is based on the finding that the A3AR is highly expressed in pathological cells, such as various tumor cell types and inflammatory cells. High A3AR expression levels are also found in peripheral blood mononuclear cells, or PBMCs, of patients with cancer, inflammatory and viral diseases. PBMCs are a critical part of the immune system required to fight infection. We believe that targeting the A3AR with synthetic and highly selective A3AR agonists, such as Piclidenoson and Namodenoson, and allosteric modulators, such as CF602, induces anti-cancer and anti-inflammatory effects. In addition, our human clinical data suggests that the A3AR is a biological marker and that high A3AR expression prior to treatment may be predictive of good patient response to our drug treatment. In fact, as a result of our research we have developed a simple blood assay to test for A3AR expression as a predictive biological marker. We have been granted a U.S. patent with respect to the intellectual property related to such assay and utilized this assay in our Phase IIb study of Piclidenoson tor the treatment of RA.
Moreover, we believe characteristics of Piclidenoson, as exhibited in our clinical studies to date, including its good safety profile, clinical activity, simple and less frequent delivery through oral administration and its low cost of production, position it well against the competition in the autoimmune-inflammatory markets, including the RA and psoriasis markets, where treatments, when available, often include injectable drugs, many of which can be highly toxic, expensive and not always effective. Furthermore, pre-clinical pharmacology studies in different experimental animal models of arthritis revealed that Piclidenoson acts as a disease modifying anti-rheumatic drug, or a DMARD, which, when coupled with its good safety profile, make it competitive in the psoriasis and RA markets. Our recent findings also demonstrate that a biological predictive marker can be utilized prior to treatment with Piclidenoson, which may allow it to be used as a personalized medicine therapeutic approach for the treatment of RA. Like Piclidenoson, Namodenoson has a good safety profile, is orally administered and has a low cost of production, which we believe positions it well in the HCC market, where only one drug, Nexavar, has been approved by the FDA.
Nevertheless, other drugs on the market, new drugs under development (including drugs that are in more advanced stages of development in comparison to our drug candidates) and additional drugs that were originally intended for other purposes, but were found effective for purposes targeted by us, may all be competitive to the current drugs in our pipeline. In fact, some of these drugs are well established and accepted among patients and physicians in their respective markets, are orally bioavailable, can be efficiently produced and marketed, and are relatively safe. None of our product candidates have been approved for sale or marketing and, to date, there have been no commercial sales of any of our product candidates.
Our research further suggests that A3AR affects pathological and normal cells differently. While specific A3AR agonists, such as Piclidenoson and Namodenoson, and allosteric modulators, such as CF602, appear to inhibit growth and induce apoptosis of cancer and inflammatory cells, normal cells are refractory, or unresponsive to the effects of these drugs. To date, the A3AR agonists have had a positive safety profile as a result of this differential effect.
We also seek to obtain technologies that complement and expand our existing technology base by entering into license agreements with academic institutions and biotechnology companies. We in-licensed the intellectual property rights to CF602 from Leiden University. Under this license agreement we are generally obligated to diligently pursue product development, make development milestone payments, pay royalties on any product sale and make payments upon the grant of sublicense rights. The scope of payments we are required to make under our in-licensing agreement is comprised of various components that are paid commensurate with the progressive development and commercialization of our drug products.
In addition to in-licensing, we have also out-licensed one of our molecules to third-parties to capitalize on the experience, capabilities and location of such third-parties. Similar to our obligations under any in-license agreements, pursuant to these out-licensing agreements, our licensees are generally obligated to diligently pursue product development, make up-front payments, make development milestone payments and pay royalties on sales. Accordingly, we expect to fund certain of our future operations through out-licensing arrangements with respect to our product candidates.
We are currently: (i) conducting preparatory work for a Phase III trial for Piclidenoson in the treatment of RA, following agreement with the EMA on our protocol design and expect to commence enrollment in the second quarter of 2017, (ii) conducting preparatory work for a Phase III trial for Piclidenoson in the treatment of psoriasis following agreement with the EMA on our protocol design and expect to commence Institutional Review Board, or IRB, submissions in the fourth quarter of 2017, (iii) conducting a Phase II study with respect to the development of Namodenoson for the treatment of HCC and anticipate completing enrollment of approximately 78 patients during the first half of 2017, (iv) conducting preparatory work for a Phase II trial of Namodenoson in the treatment of NASH, a new indication identified by us for our liver cancer drug, following approval of the study protocol by IRBs and anticipate commencing enrollment in the second quarter of 2017, and (v) conducting efficacy and safety IND enabling studies with two additional compounds that belong to the family of allosteric molecules, similar to CF602, for the treatment of sexual dysfunction.
| 29 |
Our Strategy
Our strategy is to build a fully integrated biotechnology company that discovers, in-licenses and develops an innovative and effective small molecule drug portfolio of ligands that bind to a specific therapeutic target for the treatment of autoimmune-inflammatory indications, oncology and liver diseases as well as sexual dysfunction. We continue to develop and test our existing pipeline, while also testing other indications for our existing drugs and examining, from time to time, the potential of other small molecules that may fit our platform technology of utilizing small molecules to target the A3AR. We generally focus on drugs with global market potential and we seek to create global partnerships to effectively assist us in developing our portfolio and to market our products. Our approach allows us to:
| ● | continue to advance our clinical and preclinical pipeline; |
| ● | test our products for additional indications which fit our molecules’ mechanism of action; |
| ● | identify other small molecule drugs or ligands; |
| ● | focus on our product candidates closest to realizing their potential; and |
| ● | avoid dependency on a small number of small molecules and indications. |
Using this approach, we have successfully advanced our product candidates for a number of indications into various stages of clinical development. Specific elements of our current strategy include the following:
Successful development of our existing portfolio of small molecule orally bioavailable drugs for the treatment of various diseases. We intend to continue to develop our existing portfolio of small molecule orally bioavailable drugs, both for existing targeted diseases, as well as other potential indications. Our drug development will continue to focus on autoimmune- inflammatory, oncology and liver diseases as well as sexual dysfunction. We will focus most prominently on advancing our product candidates that are in the most advanced stages, i.e., plaque psoriasis and RA with respect to Piclidenoson, and HCC and NASH with respect to Namodenoson.
Use our expertise with our platform technology to evaluate in-licensing opportunities. We continuously seek attractive product candidates and innovative technologies to in-license or acquire. We intend to focus on product candidates that would be synergistic with our A3AR expertise. We believe that by pursuing selective acquisitions of technologies in businesses that complement our own, we will be able to enhance our competitiveness and strengthen our market position. We intend to utilize our expertise in A3AR and our pharmacological expertise to validate new classes of small molecule orally bioavailable drugs. We will then seek to grow our product candidate portfolio by attempting to in-license those various candidates and to develop them for a variety of indications.
Primarily develop products that target major global markets. Our existing product candidates are almost all directed at diseases that have major global markets. Our intent is to continue to develop products that target diseases that affect significant populations using our platform technology. We believe these arrangements will allow us to share the high development cost, minimize the risk of failure and enjoy our partners’ marketing capabilities, while also enabling us to treat a more significant number of persons. We believe further that this strategy will increase the likelihood of advancing clinical development and potential commercialization of our product candidates.
Commercialize our product candidates through out-licensing arrangements. We have previously entered into two out-licensing arrangements with major pharmaceutical companies in the Far East and one distribution agreement with a growing pharmaceutical company in Canada. We intend to continue to commercialize our product candidates throughout-licensing arrangements with third parties who may perform any or all of the following tasks: completing development, securing regulatory approvals, manufacturing, marketing and sales. We do not intend to develop our own manufacturing facilities or sales forces. If appropriate, we may enter into co-development and similar arrangements with respect to any product candidate with third parties or commercialize a product candidate ourselves. We believe these arrangements will allow us to share the high development cost, minimize the risk of failure and enjoy our partners’ marketing capabilities. We believe further that this strategy will increase the likelihood of advancing clinical development and potential commercialization of our product candidates.
| 30 |
Our Product Pipeline
The table below sets forth our current pipeline of product candidates, including the target indication and status of each.
| Clinical Application/Drug | Pre-Clinical | Phase I | Phase II | Phase III | |||||
| Autoimmune-Inflammatory | |||||||||
| Rheumatoid Arthritis - Piclidenoson (1) | |||||||||
| Psoriasis - Piclidenoson (2) | |||||||||
| Oncology/Liver diseases | |||||||||
| HCC - Namodenoson(3) | |||||||||
| NASH – Namodenoson(4) | |||||||||
| Sexual Dysfunction - CF602 (5) | |||||||||
| Completed | |||||||||
| On-going | |||||||||
| Preparatory work |
| (1) | We are conducting preparatory work for a Phase III trial for Piclidenoson in the treatment of RA, following agreement with the EMA on our protocol design. |
| (2) | We are conducting preparatory work for a Phase III trial for Piclidenoson in the treatment of psoriasis following agreement with the EMA on our protocol design. |
| (3) | We are conducting a Phase II study with respect to the development of Namodenoson for the treatment of HCC with approximately 78 patients. |
| (4) | We are conducting preparatory work for a Phase II trial of Namodenoson in the treatment of NASH, a new indication identified by us for our liver cancer drug, following approval of the study protocol by IRBs. |
| (5) | We are conducting efficacy and safety IND enabling studies with two additional compounds that belong to the family of allosteric molecules, similar to CF602, for the treatment of sexual dysfunction. |
| 31 |
Piclidenoson (CF101)
Piclidenoson, our lead therapeutic product candidate, is in development for the treatment of autoimmune-inflammatory diseases. In certain of our pharmacological studies, Piclidenoson has also shown potential for development for the treatment of Crohn’s disease. Piclidenoson is a highly-selective, orally bioavailable small molecule synthetic drug, which targets the A3AR. Based on our clinical studies to date, we believe that Piclidenoson has a favorable safety profile and significant anti-inflammatory effects as a result of its capability to inhibit the production of inflammatory cytokines, such as TNF-α, IL-6 and IL-1, and chemokines, or small cytokines, such as MMPs, by signaling key proteins such as NF-кB and PKB/AKT. Overall, these up-stream events result in apoptosis of inflammatory cells. See Figure 1 below. Piclidenoson’s anti-inflammatory effect is mediated via the A3AR, which is highly expressed in inflammatory cells.
Figure 1: Piclidenoson anti-inflammatory mechanism of action
Set forth below are general descriptions of the inflammatory diseases with respect to which Piclidenoson is currently undergoing, or is in preparation for clinical trials.
Rheumatoid Arthritis: RA, is a chronic, systemic autoimmune-inflammatory disease that may affect many tissues and organs, but principally attacks flexible synovial, or joints, on both sides of the body. This symmetry helps distinguish RA from other types of arthritis, which is the general term for joint inflammation. Although the cause of RA is unknown, autoimmunity plays a pivotal role in both its chronicity and progression. The disease involves abnormal B cell–T cell interaction, which results in the release of cytokines. The cytokines signal the release of inflammatory cells. The inflammatory cells migrate from the blood into the joints and joint-lining tissue. There, the cells produce inflammatory substances that cause irritation, wearing down of cartilage, or the cushioning material at the end of bones, swelling and inflammation of the joint lining, which is caused by excess synovial fluid, the development of pannus, or fibrous tissue, in the joint, and ankylosis, or fusion of the joints. Joint inflammation is characterized by redness, warmth, swelling and pain within the joint. As the cartilage wears down, the space between the bones narrows. If the condition worsens, the bones could rub against each other. As the lining expands due to inflammation from excess fluid, it may erode the adjacent bone, resulting in bone damage. RA can also produce diffuse inflammation in the lungs, membrane around the heart, the membranes of the lungs, and white of the eye, and also nodular lesions, most common in subcutaneous tissue.
Psoriasis: Psoriasis is an autoimmune hereditary disease that affects the skin. In psoriasis, immune cells move from the dermis to the epidermis, where they stimulate keratinocytes, or skin cells, to proliferate. DNA acts as an inflammatory stimulus to stimulate receptors which produce cytokines, such as IL-1, IL-6, and TNF-α, and antimicrobial peptides. These cytokines and antimicrobial peptides signal more inflammatory cells to arrive and produce further inflammation. In other words, psoriasis occurs when the immune system overreacts and mistakes the skin cells as a pathogen, and sends out faulty signals that speed up the growth cycle of skin cells. Normally, skin cells grow gradually and flake off approximately every four weeks. New skin cells grow to replace the outer layers of the skin as they shed. But in psoriasis, new skin cells move rapidly to the surface of the skin in days rather than weeks. They build up and form thick patches called plaques.
There are five types of psoriasis: plaque, guttate, inverse, pustular and erythrodermic. The most common form, plaque psoriasis, is commonly seen as red and white hues of scaly patches appearing on the top first layer of the epidermis, or skin. In plaque psoriasis, skin rapidly accumulates at these sites, which gives it a silvery-white appearance. Plaques frequently occur on the skin of the lower back, elbows and knees, but can affect any area, including the scalp, palms of hands, soles of feet and genitals. The plaques range in size from small to large. In contrast to eczema, psoriasis is more likely to be found on the outer side of the joint. Some patients, though, have no dermatological symptoms.
Psoriasis is a chronic recurring condition that varies in severity from minor localized patches to complete body coverage. Fingernails and toenails are frequently affected, known as psoriatic nail dystrophy, and can be seen as an isolated symptom. Psoriasis can also cause inflammation of the joints, which is known as psoriatic arthritis.
| 32 |
Pre-Clinical Studies of Piclidenoson
The information below is based on the various studies conducted with Piclidenoson, including preclinical studies. All of the studies were conducted by Can-Fite and/or by Can-Fite’s partners or affiliates.
Pre-clinical studies are a set of experiments carried out in animals to show that a certain drug does not evoke toxicity. Based on the animal studies and safety data, one can approach the FDA and request permission to conduct a Phase I study in human beings.
The toxicity of Piclidenoson has been evaluated following 28-day, 90-day, six-month and nine-month good laboratory practice repeated-dose toxicity studies in male and female mice (28-day, 90-day and six-month), dogs (single-dose only), and monkeys (28-day, 90-day and nine-month). Even though the dose of Piclidenoson in these studies was escalated to an exposure that is many folds higher than the dose used in human clinical studies, no toxic side effects were identified.
Effects on cardiovascular parameters were evaluated in conscious instrumented monkeys and anesthetized dogs. These studies demonstrated no significant cardiovascular risk.
Genotoxicity studies were conducted in bacterial and mammalian mutation assays in vitro (i.e., laboratory) and in an in vivo (i.e., animal) mouse micronucleus assay. These studies were all negative, indicating no deleterious action on cellular genetic material.
Reproductive toxicology studies that we completed in mice and rabbits did not reveal evidence of negative effects on male or female fertility. In mouse teratology studies, or studies for abnormalities of physiological developments, craniofacial and skeletal abnormalities were observed at doses greater than 10 mg/kg; however, no such effects were observed at 3 mg/kg demonstrating the safety of the drug in this concentration range. Teratogenicity, or any developmental anomaly in a fetus, was not observed in rabbits given doses (greater than 13 mg/kg) that induced severe maternal toxicity in such rabbits.
Studies of P450 enzymes, or enzymes that participate in the metabolism of drugs, showed that Piclidenoson caused no P450 enzyme inhibition, or increased drug activity, or induction, or reduced drug activity. Studies carried out with radiolabeled (C14) Piclidenoson in rats showed that the drug is excreted essentially unchanged. These studies also showed that the drug is widely distributed in all body parts, except the central nervous system.
Clinical Studies of Piclidenoson
The information below is based on the various studies conducted with Piclidenoson, including clinical studies in patients with autoimmune-inflammatory and ophthalmic diseases. All of the studies were conducted by Can-Fite and/or by Can-Fite’s partners or affiliates.
Phase I Clinical Studies of Piclidenoson
Piclidenoson has been studied comprehensively in normal volunteer trials to assess safety, pharmacokinetic metabolism and food interaction. Two Phase I studies in 40 healthy volunteers, single dose and repeated dose, indicated that Piclidenoson is rapidly absorbed (reaching a maximal concentration within one to two hours) with a half-life of eight to nine hours. Some mild adverse events (principally, increased heart rate) were observed at doses higher than single doses of 10.0 mg and twice-daily doses of 5.0 mg. Such increase in heart rate was not accompanied by any change in QT intervals. The drug showed linear kinetics, in that the concentration that results from the dose is proportional to the dose and the rate of elimination of the drug is proportional to the concentration, and low inter-subject variability, meaning that the same dose of the drug does not produce large differences in pharmacological responses in different individuals. A fed-fast Phase I study (with and without food) demonstrated that food causes some attenuation in Piclidenoson absorption; accordingly Piclidenoson is administered to patients on an empty stomach in our trials. An additional Phase I study of the absorption, metabolism, excretion and mass balance of 4.0 mg (C14) Piclidenoson was conducted in six healthy male subjects and demonstrated that Piclidenoson was generally well-tolerated in this group.
Based on the findings from Phase I clinical studies, 4.0 mg BID, or twice daily, was selected as the upper limit for initial Phase II clinical trials.
| 33 |
Phase II, Phase II/III and Phase III Clinical Studies of Piclidenoson
Piclidenoson has completed eleven Phase II studies, one Phase II/III study and one Phase III study in different clinical indications including psoriasis, rheumatoid arthritis, glaucoma and DES, in approximately 1,500 patients. These studies indicate that Piclidenoson has a favorable safety profile at doses up to 4.0 mg BID for up to 32 weeks. In these studies, we did not observe a dose-response relationship between Piclidenoson and adverse events. Moreover, we did not observe any clinically significant changes in vital signs, electrocardiograms, blood chemistry or hematology.
Piclidenoson given as a standalone therapy reached the primary endpoint in Phase II clinical studies in DES however a Phase III study of Piclidenoson for DES failed to reach the primary endpoint. We have observed positive data utilizing Piclidenoson as a standalone drug in a Phase IIa clinical study in RA. In this study, we also observed a significant direct correlation between A3AR expression prior to treatment and the patients’ responses to Piclidenoson. However, we did not fully attain the primary endpoint in this study as we did not observe a significant difference in responses between Piclidenoson and the placebo (which for this study was 0.1 mg of Piclidenoson). Moreover, two Phase IIb studies in RA utilizing Piclidenoson in combination with methotrexate, a generic drug commonly used for treating RA patients, or MTX, also failed to reach the primary endpoints. Based on this data, we believe that the failures in the Phase IIb studies in RA may have been due to low A3AR expression in the MTX-treated patients. A Phase IIb of Piclidenoson given as a standalone therapy in patients with A3AR expression levels above a certain threshold reached the primary endpoint in RA in December 2013. Piclidenoson has been tested in Phase II trials to establish dose and activity (first, orally administered capsules and then tablets in formulations of 1.0, 2.0 and 4.0 mg of Piclidenoson BID) in psoriasis (moderate to severe plaque psoriasis), RA and DES (moderate to severe.) A Phase II/III study of Piclidenoson for psoriasis did not meet its primary endpoint although positive data from further analysis of the Phase II/III study suggests Piclidenoson as a potential systemic therapy for patients with moderate-severe psoriasis. In addition, a Phase II study of Piclidenoson for glaucoma showed no statistically significant differences between the Piclidenoson treated group and the placebo group in the primary endpoint of lowering intra ocular pressure.
Psoriasis: The rationale for utilizing Piclidenoson to treat psoriasis stems from our pre-clinical pharmacology studies showing that Piclidenoson acts as an anti-inflammatory agent via the inhibition of inflammatory cytokines, including TNF-α, which plays a major role in the pathogenesis of psoriasis. In addition, the A3AR is over-expressed in the tissue and PBMCs of patients with psoriasis.
We completed an exploratory Phase II trial in ten European and Israeli medical centers involving 76 patients. This study was a randomized, double-blind, placebo controlled and included four cohorts of 1.0, 2.0, and 4.0 mg of Piclidenoson and a placebo for a 12-week period. The study objectives were efficacy and safety of daily doses of Piclidenoson administered orally in patients with moderate-to-severe plaque-type psoriasis and the efficacy endpoints were improvements in both the Psoriasis Area Sensitivity Index score, or PASI score, and the Physicians’ Global Assessment score, or PGA score. We concluded that Piclidenoson met such efficacy endpoints and was safe, well tolerated and effective in ameliorating disease manifestations in these patients. The patient group receiving 2.0 mg Piclidenoson BID showed progressive improvement over the course of the 12-week study in the PGA and PASI scores. Analysis of the mean change from baseline in the PASI score at week 12 revealed a statistically significant difference between the 2.0 mg Piclidenoson BID treated group and the placebo group (P < 0.001 versus baseline and P = 0.031 versus placebo). Analysis of the PGA score revealed that 23.5% of the patients treated with the 2.0 mg Piclidenoson BID achieved a score of 0 or 1, in comparison to 0% in the placebo group (P < 0.05). The study also demonstrated linear improvement in patients in both PASI and PGA. See Figure 2. No drug-related serious adverse events were evident during the study.
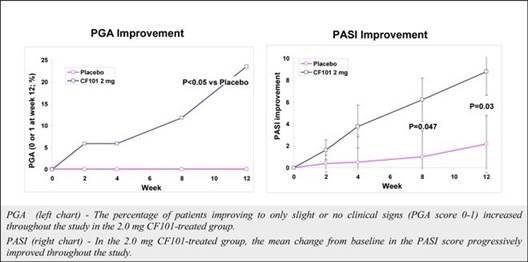
Figure 2: Psoriasis efficacy by PGA and PASI
| 34 |
Set forth below are representative pictures of a patient with plaque-type psoriasis on the upper and lower back treated with 2.0 mg Piclidenoson BID, both baseline and week 12.
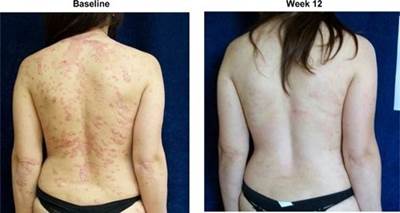
A comparison between baseline and week 12 of a patient treated with 2.0 mg CF 101
In February 2015, we completed a Phase II/III randomized, double-blind, placebo-controlled, dose-finding study of the efficacy and safety of Piclidenoson administered daily orally in patients with moderate-to-severe plaque psoriasis. This clinical trial enrolled 326 patients in 17 clinical centers in the United States, Europe and Israel, of which 103 patients were enrolled in the first study cohort and were treated for 6 months and 223 patients were enrolled in the second study cohort and were treated for 8 months. The first study cohort was comprised of three arms with patients receiving: 1 mg of Piclidenoson; 2 mg of Piclidenoson; and placebo. All patients receiving placebo were switched to either 1 mg or 2 mg of Piclidenoson after 12 weeks. Based on a positive safety and efficacy interim analysis of the first 103 patients who completed 24 weeks of treatment in the trial, we decided to continue patient enrollment for the second stage of the study and the study protocol was amended to extend the Piclidenoson 2.0 mg BID and placebo administration for a period of 32 weeks. The positive clinical effects of the Piclidenoson 2.0 mg BID dose relative to a placebo were observed in a variety of standard psoriasis assessment parameters, including PASI 75 and PGA scores, with the responses accumulating steadily over the 24-week treatment period.
In March 2015, we announced the study did not meet its primary endpoint of a statistically significant improvement in the PASI 75 score relative to placebo after 12 weeks of treatment. Further analysis of the entire study period revealed that by 32 weeks of treatment with Piclidenoson, 33% of the patients achieved PASI 75 while the mean percent of improvement in PASI score was 57% (p<0.001). This was a statistically significant cumulative and linear improvement during weeks 16 to 32. Most significantly, by week 32 of the study, 20% of the study patients reached PASI 90, a result demonstrating a response rate of 90% clearing of skin lesions. PASI 90 is one of the most stringent and difficult to meet clinical endpoints for measuring responses to psoriasis treatments. Moreover, the PASI 90 subset analysis further suggests a higher and significant (p=0.026) Piclidenoson response rate of 27% among patients previously untreated with systemic psoriasis therapy compared to patients pre-treated with systemic drugs. We believe this presents the opportunity that Piclidenoson can be developed as a first-line systemic therapy for patients with moderate-severe psoriasis and for patients who do not want to be treated with the current systemic drugs due to safety issues.
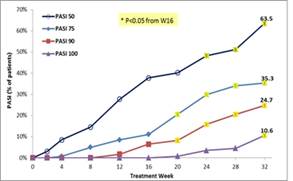
Figure 3: Linear Effect of Piclidenoson on PASI Scores through 32 Weeks of Treatment
| 35 |
We are currently planning to commence a Phase III trial for Piclidenoson for the treatment of psoriasis in 2017. In the fourth quarter of 2016, we reached an agreement with the EMA on the final design of a global pivotal Phase III trial. The planned Phase III trial is to be a randomized, double-blind, placebo- and active-controlled study that will investigate the efficacy and safety of daily Piclidenoson administered orally as compared to placebo as its primary endpoint and as compared to apremilast (Otezla®) as its secondary endpoint in approximately 400 patients with moderate-to-severe plaque psoriasis. Medication is to be taken orally twice daily for 32 weeks in a double-blinded fashion. The primary end point will be the proportion of subjects who achieve a PASI score response of ≥75% (PASI 75) vs. placebo at week 16. The secondary endpoints will include non-inferiority to Otezla® on week 32 and efficacy and safety data for CF101 through the extension period of up to 48 weeks of treatment. Patients will be selected to the study based on over expression of the A3AR biomarker. We plan to submit our clinical protocol to IRBs in the fourth quarter of 2017.
Rheumatoid Arthritis: We conducted a Phase IIa blinded to dose study in 74 patients with RA, randomized to receive Piclidenoson as a monotherapy in one of three doses—0.1 mg, 1.0 mg and 4.0 mg. The primary efficacy endpoint was ACR20 response at week 12, a criterion determined by the American College of Rheumatology that reflects 20% improvement in inflammation parameters. The study data revealed maximal response at the 1.0 mg group, showing 55.6% with ACR20, 33.3% with 50% improvement, or ACR50, and 11.5% with 70% improvement, or ACR70. Piclidenoson administered BID for 12 weeks resulted in improvement in signs and symptoms of RA and was safe and well-tolerated. See Figure 4. Studies in the United States were conducted pursuant to an open IND which was received by the FDA in 2005.
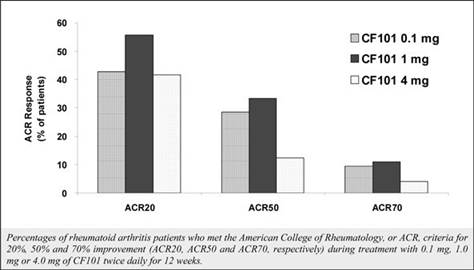
Figure 4: Rheumatoid Arthritis efficacy by ACR
| 36 |
Subsequently, two Phase IIb studies with Piclidenoson in combination with MTX were conducted. The study protocols were multicenter, randomized, double-blind, placebo-controlled, parallel-group and dose-finding to determine the safety and efficacy of daily Piclidenoson administered orally when added to weekly MTX in patients with active RA. The objectives of both studies were improvement in ACR20, ACR50, ACR70 and DAS28, or the Disease Activity Score of 28 Joints, and EULAR, or the European League Against Rheumatism, response criteria, as well as a positive safety profile. The trials’ primary endpoints were both ACR20.
The first Phase IIb trial showed that the combined treatment had an excellent safety profile, but no significant ACR20 response was observed between the RA group treated with Piclidenoson and MTX and the group treated with MTX alone (the placebo group). However, the ACR50, ACR70 and the EULAR Good Values in the combined treatment group were higher than those of the MTX placebo group. The study also indicated that the 1.0 mg Piclidenoson dose was the most favorable dose, i.e., the dose yielded the highest ACR50 and EULAR Good Values as compared to the MTX placebo group. The most commonly reported adverse events in this study included nausea, dizziness, headache and common bacterial and viral infections and infestations.
Following a decision of our Clinical Advisory Board in October 2007, an additional Phase IIb study was initiated. This study was conducted in medical centers in Europe and Israel and included 230 patients who received the drug orally BID (0.1 and 1.0 mg Piclidenoson tablets plus MTX versus a placebo, which was MTX alone) for 12 weeks. On April 30, 2009, we published preliminary results of the Phase IIb study, which were later confirmed as the final results, also indicating that the study’s objectives were not achieved. The most commonly reported adverse events in this study included nausea, myalgia and dizziness.
The two Phase IIb studies failed to achieve the primary endpoint of ACR20. A cross study analysis of the three RA clinical studies revealed that in the first Phase IIa study, where Piclidenoson had been administered as a standalone drug, A3AR had been over-expressed in the patients’ PBMCs prior to Piclidenoson treatment, whereas A3AR had not been over-expressed in the Phase IIb patient population. We believe, based on the foregoing data, that there may be a direct and statistically significant correlation between A3AR over-expression at baseline and patients’ response to Piclidenoson, and that Piclidenoson should be administered as a standalone drug and not in combination with MTX. Furthermore, the correlation between A3AR expression levels prior to treatment and patients’ response to the drug suggest that the A3AR may be a predictive biomarker to be analyzed prior to Piclidenoson treatment. See Figures 5 and 6.
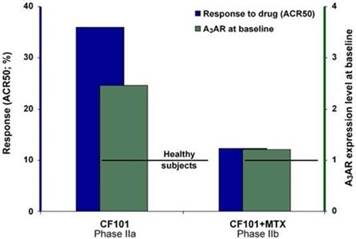
Figure 5: Direct correlation between A3AR at baseline and response to Piclidenoson
| 37 |
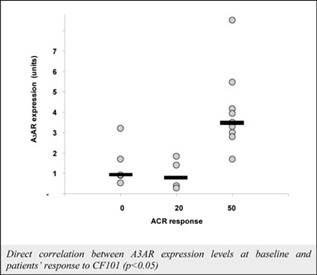
Figure 6: Direct correlation between A3AR at baseline and response to Piclidenoson
Based on the results of the two Phase IIb studies, we conducted an additional Phase IIb clinical study with Piclidenoson as a stand-alone, monotherapy treatment and not in combination with MTX. The trial was a 12-week multicenter, randomized, double-blind, placebo-controlled, parallel-group study involving 79 patients to determine the safety and efficacy of Piclidenoson administered orally daily in patients with active RA and elevated baseline expression levels of the A3AR in PBMCs. Enrolled patients had high baseline A3AR biomarker expression (determined at 1.5-fold over a predetermined age-matched standard). This selection criteria was made following the findings during previous Phase IIa and IIb RA studies showing a positive correlation between A3AR expression at baseline and patients’ response to the drug, potentially rendering A3AR expression as a predictive biomarker. The primary objectives of this study were to determine the efficacy of oral Piclidenoson when administered daily as a standalone treatment for 12 weeks to patients with active RA and elevated baseline expression levels of the A3AR in the patients’ PBMCs, in comparison to a placebo treatment, and to assess the safety of daily oral Piclidenoson under the circumstances of the trial. In December 2013, we announced the results of the study in which Piclidenoson met all primary efficacy endpoints, showing statistically significant superiority over placebo in reducing signs and symptoms of RA as compared to the placebo. The treatment had an ACR20 response rate of 49% for Piclidenoson compared to 25% for placebo (p=0.035), an ACR50 response rate of 19% for Piclidenoson compared to 9% for placebo, and an ACR70 response rate of 11% for Piclidenoson arm compared to 3% for placebo. Similar to our observations in the previously reported Piclidenoson psoriasis trials, the response of patients with RA was cumulative over time, suggesting a consistent anti-inflammatory effect of Piclidenoson. Moreover, half of the RA patients treated with Piclidenoson showed clinically meaningful improvement. Piclidenoson was very well-tolerated and showed no evidence of immunosuppression, and there were no severe treatment-emergent adverse events during the study. A subgroup analysis of 16 patients with no prior systemic therapy showed a dramatic increase in the response showing ACR20 of 75%, ACR50 50%, and ACR70 50%. We believe this may be related to the fact that in this patient population there is a full receptor expression since they had not been treated earlier with any systemic drugs.
| 38 |
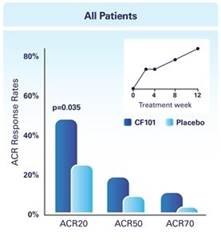 |
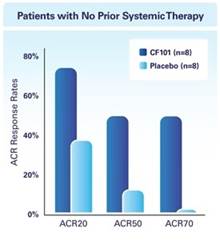 |
Figure 7: ACR response data –Rheumatoid Arthritis phase IIb
We plan to commence in the second quarter of 2017 ACRobat, our Phase III trial of Piclidenoson for RA. Piclidenoson is being developed as a first line therapy and replacement for the current standard of care, MTX, the most widely used drug for RA. The planned trial will be a randomized, double-blind, active and placebo-controlled Phase III trial to establish non-inferiority of Piclidenoson versus MTX, conducted in approximately 500 patients worldwide. The primary endpoint of ACRobat is low disease activity after 12 weeks of treatment in patients dosed with Piclidenoson compared to those dosed with MTX. Piclidenoson at 1 mg and 2 mg, or placebo, will be administered twice daily, and MTX or placebo will be administered once weekly. The total study duration will be 24 weeks in order to provide more data on long term efficacy and safety.
DES: DES is an eye disease caused by eye dryness, which, in turn, is caused by either decreased tear production or increased tear film evaporation. A Phase II study in DES was conducted by Can-Fite after discovering that patients in the Phase IIa study for another condition also experienced improvement in DES symptoms. The results of the Phase II trial demonstrated the ability of Piclidenoson to improve signs of ocular surface inflammation of the patients studied. Following positive results in the Phase II study, we initiated a Phase III DES trial, under an IND with the FDA which was conducted by OphthaliX in the United States, Europe and Israel. The randomized, double-masked Phase III clinical trial enrolled 237 patients with moderate-to-severe DES who were randomized to receive two oral doses of Piclidenoson (0.1 and 1.0 mg) and a placebo, for a period of 24 weeks. The primary efficacy endpoint was complete clearing of corneal staining. In December 2013, we announced the results of this Phase III study of Piclidenoson for the treatment of DES. In the study, Piclidenoson did not meet the primary efficacy endpoint of complete clearing of corneal staining, nor the secondary efficacy endpoints. Nonetheless, Piclidenoson was found to be well tolerated. In 2014 we decided to end the development of Piclidenoson for the DES indication. This decision was based on a lack of correlation between patients' response to Piclidenoson and over-expression of the drug target, the A3 adenosine receptor in this patient population.
Glaucoma: Glaucoma is an eye disease in which the optic nerve is damaged. This optic nerve damage involves loss of retinal ganglion cells, or neurons located near the inner surface of the retina, in a characteristic pattern. There are many different subtypes of glaucoma, but they can all be considered to be a type of optic neuropathy. Raised intraocular pressure, or IOP, is the most important and only modifiable risk factor for glaucoma. However, some individuals may have high IOP for years and never develop optic nerve damage. This is known as ocular hypertension. Others may develop optic nerve damage at a relatively low IOP, and, thus, glaucoma. Untreated glaucoma can lead to permanent damage of the optic nerve and resultant visual field loss, which over time can progress to blindness.
A Phase II clinical trial of Piclidenoson for the treatment of glaucoma was conducted by OphthaliX. The randomized, double-masked, placebo-controlled, parallel-group Phase II clinical trial was designed to evaluate the safety and efficacy of Piclidenoson when administered orally twice daily for up to 16 weeks in patients with elevated IOP. A total of 89 patients were enrolled in the study. The study was conducted with two cohorts. In the first cohort, treatment was randomized in a 3:1 ratio of 1.0 mg Piclidenoson to placebo. In the second cohort, which was also randomized in a 3:1 Piclidenoson to placebo ratio, the Piclidenoson dose was increased to 2.0 mg. In July 2016, top line results were announced. In this trial, no statistically significant differences were found between the Piclidenoson treated group and the placebo group in the primary endpoint of lowering IOP. Piclidenoson was found to have a favorable safety profile and was well tolerated. Based on these overall results OphthaliX saw no immediate path forward in glaucoma and has since ceased research and development operations.
| 39 |
Additional Developments with Piclidenoson
Osteoarthritis
According to the Arthritis Foundation, OA is the most common arthritic disease. Currently, there is a shortage of effective drugs for treating OA patients. Piclidenoson has induced a significant anti-inflammatory effect in experimental animal models with respect to the treatment of OA and, as such, we are currently preparing for a Phase II study. We have not yet filed an IND for this indication as Piclidenoson for the treatment of OA is not currently being clinically tested in the United States and there are no near-term plans to do so.
Crohn’s Disease
Crohn’s disease is an inflammatory bowel disease that may affect any portion of the gastrointestinal tract, causing a wide variety of symptoms. It primarily causes abdominal pain, diarrhea, vomiting and weight loss, however, it may also cause complications outside the gastrointestinal tract, such as skin rashes, arthritis, inflammation of the eye, tiredness and lack of concentration. Pre-clinical pharmacology studies that we have conducted demonstrated the efficacy of Piclidenoson for the treatment of Crohn’s disease. We do not presently have plans for the treatment of Crohn’s disease.
Namodenoson (CF102)
Namodenoson is our second drug candidate and is under development for the treatment of HCC, HCV, non-alcoholic fatty liver disease, or NAFLD, the precursor to non-alcoholic steatohepatitis, or NASH. Namodenoson is also a small, orally bioavailable molecule, and an A3AR agonist, with high affinity and selectivity to the A3AR. In comparison to the expression in adjacent normal liver tissue, the A3AR is over-expressed in tumor tissues of patients with HCC, and the over-expression is also reflected in the patients’ PBMCs. A3AR over-expression in the patients’ tumor cells and PBMCs is attributed to high expression of certain A3AR transcription factors. The binding of Namodenoson to the A3AR results in down-regulation, or a decrease in the quantity of a cellular component, such as the number of receptors on a cell’s surface, of certain A3AR transcription factors. Our studies have shown that this down-regulation leads to apoptosis of HCC cells. In our pre-clinical and clinical studies, Namodenoson demonstrated anti-cancer, anti-viral and liver protective effects. As a result, we believe that Namodenoson can be used to treat a variety of oncological and liver-related diseases and viruses.
In February 2012, the FDA granted an orphan drug status for the active moiety, or the part of the drug that is responsible for the physiological or pharmacological action of the drug substance, of Namodenoson for the treatment of HCC. Subsequently, in October 2015, the EMA granted Namodenoson orphan drug designation for the treatment of HCC.
An orphan drug designation is a special designation for drug approval and marketing. The special designation is granted to companies that develop a given drug for unique populations and for incurable and relatively rare diseases. The FDA orphan drug designation program provides orphan status to drugs and biologics which are intended for the safe and effective treatment, diagnosis or prevention of rare diseases or disorders that affect fewer than 200,000 people in the United States and in the EU not more than 5 per 10,000. Orphan drug designations have enabled companies to achieve medical breakthroughs that may not have otherwise been achieved due to the economics of drug research and development as this status lessens some of the regulatory burdens, for approval, including statistical requirements for efficacy, safety and stability, in an effort to maintain development momentum. Orphan drug designation also results in additional marketing exclusivity and could result in certain financial incentives.
In September 2015, the FDA granted Fast Track designation to Namodenoson as a second line treatment to improve survival for patients with advanced HCC who have previously received Nexavar (sorafenib). Fast Track, aimed at getting important new drugs that meet an unmet need to patients earlier, is expected to expedite the development of Namodenoson. Drugs that receive Fast Track designation benefit from more frequent meetings and communications with the FDA to review the drug's development plan to support approval. It also allows the Company to submit parts of the NDA on a rolling basis for review as data becomes available.
Israel's Ministry of Health has previously approved Namodenoson for Compassionate Use for HCC.
| 40 |
Set forth below are general descriptions of the diseases with respect to which Namodenoson has underwent or is currently undergoing or being prepared for clinical trials.
HCC: HCC is an oncological disease characterized by malignant tumors that grow on the surface or inside of the liver. This type of tumor is refractory to chemotherapy and to other anti-cancer agents. HCC, like any other cancer, develops when there is a mutation to the cellular machinery that causes the cell to replicate at a higher rate and/or results in the cell avoiding apoptosis. Chronic infections of Hepatitis B and/or C can aid the development of HCC by repeatedly causing the body’s own immune system to attack the liver cells, some of which are infected by the virus. While this constant cycle of damage followed by repair can lead to mistakes during repair which in turn lead to carcinogenesis, this hypothesis is more applicable, at present, to HCV. Chronic HCV causes HCC through cirrhosis. In chronic Hepatitis B, however, the integration of the virus into infected cells can directly induce a non-cirrhotic liver to develop HCC. Alternatively, repeated consumption of large amounts of ethanol can have a similar effect.
Hepatitis C: HCV is an infectious disease affecting primarily the liver, caused by the Hepatitis C virus. The infection is often asymptomatic, but chronic infection can lead to scarring of the liver and ultimately to cirrhosis, which is generally apparent after many years, and chronic liver disease. The virus also increases the chance for HCC development. In some cases, those with cirrhosis will develop liver failure, liver cancer or life-threatening esophageal and gastric varices, or dilated submucosal veins, which can be life-threatening. HCV is spread primarily by blood-to-blood contact often associated with intravenous drug use, poorly sterilized medical equipment, transfusions, and sexual intercourse.
NAFLD/NASH: NASH, also called "fatty liver", is a condition in which fat builds up inside the liver causing inflammation. Prior to the presence of inflammation, the disease is simply referred to as non-alcoholic fatty liver disease, or NAFLD, the most common form of liver disorder in the United States. The accumulation of macroglobular fat inside the liver causes oxidative stress that reduces the efficiency of the liver and can lead to increased liver enzymes such as alanine aminotransferase and aspartate aminotransferase. Loss of liver efficiency and oxidative stress leads to inflammation, liver cell ballooning, and the development of NASH. Prolonged inflammation results in cirrhosis (scar tissue), liver failure, or liver cancer. There are currently no drugs approved for the treatment of NASH.
Pre-Clinical Studies of Namodenoson
We conducted several pre-clinical studies, including studies of toxicity. The results indicated that Namodenoson was well- tolerated with no adverse effects. In these studies, we evaluated the toxicity, stability, metabolism and other safety parameters of Namodenoson at doses much higher than the doses that we currently administer to humans in our clinical trials of Namodenoson.
In pre-clinical pharmacology studies, Namodenoson inhibited the growth of HCC via the induction of tumor cell apoptosis. In addition, in collaboration with leading virology labs, we observed that Namodenoson inhibited viral replication of HCV through the down-regulation of viral proteins. Both of these findings served as a basis to further explore development of this drug for HCC and HCV.
Moreover, our pre-clinical studies demonstrated that Namodenoson acted to stimulate liver regeneration after partial hepatectomy, or removal of a part of the liver. The studies showed that Namodenoson protected the liver against ischemic reperfusion manifested by a statistically significant (p<0.05) reduction in key liver enzymes, SGOT and SGPT. In addition, in studies where partial liver hepatectomy was conducted, a 45% increase in the regeneration rate of the remaining liver was observed after Namodenoson treatment, compared to placebo which regenerated only by 24%.
In a preclinical study, Namodenoson also revealed its capability to improve liver pathology in a NAFLD /diabetes animal model of NASH. The data showed:
| ● | Namodenoson had a statistically significant reduction in NAFLD activity score compared to placebo; |
| ● | Namodenoson reduced liver-to-body weight compared to placebo; |
| ● | Representative photomicrographs of H&E-stained liver sections showed improved pathology in animals receiving Namodenoson vs. placebo; |
| 41 |
| ● | Namodenoson decreased plasma ALT and triglycerides levels in the livers of NASH-model compared to placebo; |
| ● | Liver sections from the placebo group exhibited severe micro- and macrovesicular fat deposits, hepatocellular ballooning and inflammatory cell infiltration, whereas the Namodenoson treated group showed a significant decrease in steatosis, ballooning and lobular inflammation compared to the placebo group. |
In further pre-clinical studies which evaluated the effects of Namodenoson on fibrogenic hepatic stellate cells, preclinical data show Namodenoson inhibited, in a dose dependent manner, the growth and proliferation of the liver fibrosis cells. This outcome suggests the anti-fibrotic effect of the drug and supports its development as an agent to combat NAFLD, the precursor to NASH.
Most recently, preclinical studies in a mouse model of liver fibrosis demonstrated the anti-fibrotic effects of Namodenoson. The Namodenoson treated group exhibited normal liver under macroscopic view, no accumulation of fluid (ascites), a low fibrosis profile, and lower serum levels of transaminases as compared to the control group. In addition, liver protein extracts and mRNA for the alpha smooth muscle actin showed a significant anti-fibrotic effect in the Namodenoson treated group as compared to the control group.
Clinical Studies of Namodenoson
The information discussed below is based on the various studies conducted by Can-Fite with Namodenoson, including clinical studies in patients with oncological and liver-related diseases and viruses.
Phase I Clinical Study
Namodenoson completed a Phase I double-blind, randomized, placebo-controlled, ascending single dose trial to evaluate the safety, tolerability, and pharmacokinetics of orally administered Namodenoson in healthy volunteers. The study was conducted in the United States under an open IND. Namodenoson was found to be safe and well-tolerated with a half-life time of 12 hours. See Figure 10.
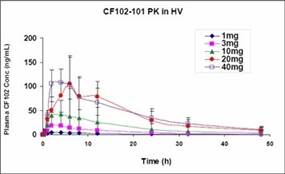
Figure 10. Namodenoson Pharmacokinetic profile
| 42 |
Phase I/II Clinical Study
HCC/HCV
Namodenoson completed two Phase I/II studies in Israel, one in patients with HCC and another in patients with HCV. The HCC Phase I/II study was an open-label, dose-escalation study evaluating the safety, tolerability, pharmacokinetics and pharmacodynamics of orally administered Namodenoson in patients with advanced HCC. The primary objectives of the study were to determine the safety and tolerability, dose-limiting toxicities, maximum tolerated dose, and recommended Phase II dose of orally administered Namodenoson in patients with advanced HCC; and to assess the repeat-dose pharmacokinetics behavior of Namodenoson in those patients. The secondary objectives were to document any observed therapeutic effect of Namodenoson in patients with HCC and to evaluate the relationship between PBMCs and the A3AR expression at baseline, as a biomarker, and the effects of Namodenoson in patients with HCC. The study included 18 patients, nine of which were also carriers of HCV. The initial dose of Namodenoson was 1.0 mg BID, with planned dose escalations in subsequent cohorts to 5.0 and 25.0 mg BID. This Phase I/II study achieved its objectives, showing a good safety profile, or no material differences versus a placebo with respect to observed and patient-indicated side effects, for Namodenoson and a linear pharmacokinetic drug profile, with no dose-limiting toxicities at any dose level. The median overall survival time for the patients in this study was 7.8 months, which is encouraging data considering that (i) 67% of the patient population in the study had previously progressed on Nexavar, produced by Onyx Pharmaceuticals and Bayer, and that Namodenoson was a second line therapy for these patients and (ii) 28% of the patient population were Child-Pugh Class B patients (patients classified on the Child Pugh scoring system for chronic liver disease as having significantly impaired liver function) whose overall survival time is usually 3.5 to 5.5 months. Accordingly, we may also consider Namodenoson as a drug to be developed for this patient sub-population of Child-Pugh Class B patients. Namodenoson had no adverse effect on routine measures of liver function over a six-month period in 12 patients treated for at least that duration. These findings are consistent with our pre-clinical Namodenoson data which demonstrated a protective effect on normal liver tissue in an experimental model of liver inflammation. As such, Namodenoson may potentially be a safer alternative to patients with cirrhosis and/or hepatic impairment. The study also demonstrated a direct relationship between A3AR expression at baseline and patients’ response to Namodenoson, suggesting A3AR as a predictive biological marker. We also observed a decrease in the viral load of seven out of nine patients who were also carriers of HCV. The most commonly reported adverse events included loss of appetite, ascites, nausea, diarrhea, constipation and pain. However, many of these events are expected in a population of patients with advanced HCC. The most frequently reported drug-related adverse events included diarrhea, fatigue, loss of appetite, pain and weakness.
Our second Phase I/II study was a randomized, double-blind, placebo-controlled, dose-escalation study evaluating the safety, tolerability, biological activity, and pharmacokinetics of orally administered Namodenoson in 32 subjects with chronic HCV genotype 1. Eligible subjects were assigned in a 3:1 ratio (eight subjects in each cohort) to receive QD or BID treatment (1.0, 5.0 and 25.0 mg of Namodenoson) for 15 days with oral Namodenoson or with a placebo. Dose escalation occurred in four sequential cohorts. The study’s primary objectives were to determine the safety and tolerability of orally administered Namodenoson in patients with chronic HCV genotype 1, to assess the effects on HCV load during 15 days of treatment with Namodenoson and to assess the repeat-dose pharmacokinetic behavior of Namodenoson under the conditions of this trial. The secondary objective of this trial was to perform an exploratory evaluation of the relationship between A3AR in PBMCs at baseline and the clinical effects of Namodenoson on the study’s patients. Following the decrease in HCV load that had been observed in HCV patients treated with Namodenoson in the parallel HCC study and the good safety profile of Namodenoson, we received Israeli IRB, approval to extend the treatment period of the Phase I/II in patients with HCV to four months with the 1.0 mg dose vs. the placebo. The results of this Phase I/II HCV study demonstrated safety and a linear pharmacokinetic drug profile, however, no significant decrease in the viral load was observed. Notwithstanding, we did observe in the parallel HCC study that seven out of the nine patients with both HCC and HCV experienced a decrease in viral load and that these seven patients were treated with higher Namodenoson dosages than what was administered to the patients with chronic HCV genotype 1 only, and not HCC, possibly explaining the difference in results. The most commonly reported adverse events included loss of appetite, ascites, nausea, diarrhea, constipation and pain. However, many of these events are expected in a population of patients with advanced HCV. The most frequently reported drug-related adverse events included diarrhea, fatigue, loss of appetite, pain and weakness.
We are conducting a Phase II study in HCC patients. In January 2013, as part of our preparatory work for such study, we announced that we believe that the optimal drug dose for the upcoming study is Namodenoson 25.0 mg. This dose was found to be the most effective dose out of the three dosages tested (1.0 mg, 5.0 mg and 25.0 mg) in the previous Phase I/II study. We filed a patent application protecting such optimal dose of Namodenoson for HCC. A publication summarizing the results of the Phase I/II study was published in “The Oncologist”, a leading oncology scientific journal. We also highlighted that one patient has been treated with Namodenoson for over five years. Also as part of the Phase II study, we plan to examine the viral load of HCC patients who are also infected with HCV. If we observe a decrease in the viral load in the HCV sub-population during this forthcoming study, we intend to commence a separate Phase II study for the HCV indication.
The Phase II study is a randomized, double-blind, placebo controlled trial conducted in the U.S., Europe and Israel with an estimated 78 patients to be enrolled. Namodenoson is being evaluated for efficacy and safety as a second-line treatment for advanced HCC in subjects with Child-Pugh B who failed Nexavar as a first line treatment. The primary endpoint of the study is overall patient survival. In March 2014, the study protocol was approved by the IRB at the Rabin Medical Center in Israel and in December 2014 we dosed the first patient at the study’s Israeli site. We expect to complete patient enrollment in the first half of 2017.
| 43 |
NAFLD/NASH
We are planning to commence a Phase II trial of Namodenoson in the treatment of NAFLD/NASH and have received approval to commence enrollment from the Institutional Review Boards of Hadassah Medical Center and Rabin Medical Center, two leading medical institutions in Israel where the study will be conducted.
The approved clinical protocol is a Phase II multicenter, randomized, double-blinded, placebo-controlled, dose-finding study of the efficacy and safety of Namodenoson in the treatment of NAFLD/NASH. Approximately 60 patients with NAFLD, with or without NASH, will be enrolled in three arms, including two different dosages of Namodenoson and a placebo, given via oral tablets twice daily. The study's primary endpoints will be percent change from baseline in liver triglyceride (fat) concentration measured by nuclear magnetic resonance spectroscopy (NMRS) and safety. Secondary endpoints to be evaluated are the effects of Namodenoson on metabolic abnormalities in subjects with NAFLD, including body weight, waist circumference, serum triglyceride and high-density lipoprotein cholesterol levels, and serum liver transaminase. In addition, an assessment of the pharmacokinetics (PK) of Namodenoson and the A3 adenosine receptor (A3AR) biomarker will be evaluated prior to treatment and its correlation to patients' response to the drug will be analyzed upon study conclusion. Furthermore, the exploratory objective of this study is to evaluate the effects of Namodenoson on relevant biomarkers, such as adiponectin, leptin, C-reactive protein (CRP), and liver stiffness as determined by Fibroscan.
Additional Developments with Namodenoson
JC Virus
In April 2011, we announced that, in laboratory study, Namodenoson inhibited the reproduction of the JC virus, a type of polyomavirus, which is dormant in approximately 70% to 90% of the world population. However, in patients treated with biological drugs, including monoclonal antibody therapeutics, such as anti-TNFs or anti-CD20, JC virus replication may occur, resulting in development of progressive multifocal leukoencephalopathy, or PML, which is characterized by progressive damage or inflammation of the white matter of the brain and, eventually, death. The ability of Namodenoson to suppress the JC Virus culture, as indicated in the laboratory study, may indicate that it may be used for the treatment of PML as a combination therapy with biological drugs. As Namodenoson is already in various stages of clinical development for other indications, its efficacy for this new application may be tested in clinical trials.
CF602
The allosteric modulator, CF602, is our third drug candidate in its pipeline. CF602 is an orally bioavailable small molecule, which enhances the affinity of the natural ligand, adenosine, to its A3AR. The advantage of this molecule is its capability to target specific areas where adenosine levels are increased. Normal body cells and tissues are refractory to allosteric modulators. This approach complements the basic platform technology of Can-Fite, utilizing the Gi coupled protein A3AR as a potent target in inflammatory diseases. CF602 has demonstrated proof of concept for anti-inflammatory activity in in vitro and in vivo studies performed by us.
During clinical studies conducted with our product candidates, other than CF602, patients suffering from sexual dysfunction reported that they returned to normal functioning following the treatment with such drugs. We believe that these findings are correlated with our platform technology, which is the targeting of the A3AR. Adenosine, like nitric oxide, is a potent and short-lived vaso-relaxant that functions via intracellular signaling (in particular, through cAMP) to promote smooth muscle relaxation. Recent studies conducted by others show that adenosine functions to relax the corpus cavernosum and thereby promote penile erection.
CF602 was tested in an experimental animal model of diabetic rats, which similar to diabetic patients, suffer from sexual dysfunction. Erectile dysfunction was assessed by monitoring the ratio between intra-cavernosal pressure (ICP) and mean arterial pressure (MAP) as a physiological index of erectile function. The ICP/MAP for the CF602 treated group improved by 118% over the placebo group. This data is similar to that achieved earlier by sildenafil (Viagra) in preclinical studies. In addition, treatment with CF602 reversed smooth muscle and endothelial damage, in a dose dependent manner, leading to the improvement in erectile dysfunction.
Further studies of CF602 have revealed that CF602 restores the impaired vascular endothelial growth factor system in the penis of diabetes mellitus rats, thereby inducing an increase in nitric oxide resulting in significant improvement of penile erection compared to placebo. This mechanism of action is similar to that of sildenafil, with CF602 demonstrating effects on erection superior to that demonstrated by sildenafil in animal studies. Among the most important factors to affect erectile function is nitric oxide which is released by endothelial cells that line the corpus cavernosum and control smooth muscle relaxation and vascular inflow. It has been well established that release of nitric oxide is diminished in diabetes.
| 44 |
In addition, CF602 induced a dose-dependent, linear effect in a diabetic mellitus rat model after treatment with one single dose of CF602. One hour after dosing, sexual function was measured. Statistically significant full recovery from erectile dysfunction took place in rats treated with a 500 µ/kg dose.
According to the American Diabetes Association, approximately 30 million children and adults have diabetes mellitus in the U.S. It is estimated that 35-75% of men with diabetes mellitus suffer from erectile dysfunction.
In November 2016, a Notice of Allowance was granted to us by the U.S. Patent and Trademark Office for our patent covering A3AR ligands for use in the treatment of erectile dysfunction. The patent addresses methods for treating erectile dysfunction with different A3AR ligands including our erectile dysfunction drug candidate, CF602. With this new broader patent protection, we made a strategic decision to investigate additional compounds, owned by us, for the most effective and safest profile in this indication. As such, we postponed our planned IND submission for this indication and are currently conducting efficacy and safety IND enabling studies with two additional compounds that belong to the family of allosteric molecules, similar to CF602, for the treatment of sexual dysfunction.
Commercial Biomarker Test
In March 2015, we completed the development of a commercial predictive biomarker blood test kit for A3AR. The biomarker test can be used at any molecular biology lab, where a small blood sample from a prospective patient would be tested and within just a few hours, results indicate if the patient would benefit from treatment with our drugs, which are currently in clinical trials for rheumatoid arthritis, psoriasis, and liver cancer.
The U.S. Patent and Trademark Office previously issued to us a patent for the utilization of A3AR as a biomarker to predict patient response to its drug Piclidenoson in autoimmune inflammatory indications.
In-Licensing Agreements
The following is a summary description of our in-licensing agreement with Leiden University. Our license with NIH expired in June 2015 with the expiration of certain patents. The description provided below does not purport to be complete and is qualified in their entirety by the complete agreement, which is attached as an exhibit to this Annual Report on Form 20-F.
Leiden University Agreements
On November 2, 2009, we entered into a license agreement, or the Leiden University Agreement, with Leiden University. Leiden University is affiliated with the NIH and is the joint owner with the NIH of the patents licensed pursuant to the Leiden University Agreement. The Leiden University Agreement grants an exclusive license for the use of the patents of several compounds, including CF602, that comprise certain allosteric compound drugs, and for the use, sale, production and distribution of products derived from such patents in the territory, i.e., China and certain countries in Europe (Austria, Belgium, Denmark, France, Germany, Italy, Spain, Sweden, Switzerland, Holland and England). Subject to certain conditions, we may sublicense the Leiden University Agreement. However, the U.S. government has an irrevocable, royalty-free, paid-up right to practice the patent rights throughout the territory on behalf of itself or any foreign government or international organization pursuant to any existing or future treaty or agreement to which the U.S. government is a signatory and the U.S. government may require us to grant sublicenses when necessary to fulfill health or safety needs.
Pursuant to the Leiden University Agreement, we are committed to make the following payments: (i) a one-time concession commission of 25,000 Euros; (ii) annual royalties of 10,000 Euros until clinical trials commence; (iii) 2% to 3% of net sales value, as defined in the Leiden University Agreement, received by us; (iv) royalties of up to 850,000 Euros based on certain progress milestones in the clinical stages of the products which are the subject of the patent under the Leiden University Agreement; and (v) if we sublicense the agreement, we will provide Leiden University royalties at a rate of 2-3% of net sales value, as defined in the Leiden University Agreement, and 10% of certain consideration received for granting the sublicense. In the event that we transfer to a transferee the aspect of our business involving the Leiden University Agreement, we must pay to Leiden University an assignment royalty of 10% of the consideration received for the transfer of the agreement. However, a merger, consolidation or any other change in ownership will not be viewed as an assignment of the agreement. In addition, we have agreed to bear all costs associated with the prosecution of the patents and patent applications to which we are granted a license under the Leiden University Agreement. As of December 31, 2016, we have paid approximately 95,000 Euros in royalties to Leiden University in connection with the Leiden University Agreement.
The Leiden University Agreement expires when the last of the patents expires in each country of the territory, unless earlier terminated in accordance with the terms of the Leiden University Agreement. The last of such patents is set to expire on 2027. The termination rights of the parties include, but are not limited to, (i) the non-defaulting party’s right to terminate if the defaulting party does not cure within 90 days of written notice identifying the default and requesting remedy of the same; and (ii) Leiden University’s right to terminate if we become insolvent, have a receiver appointed over our assets or initiate a winding-up. In addition, Leiden University may terminate the agreement when it is determined, in consultation with NIH, that termination is necessary to alleviate health and safety needs and certain other similar circumstances.
| 45 |
Out-Licensing and Distribution Agreements
The following are summary descriptions of certain out-licensing and distribution agreements to which we are a party. The descriptions provided below do not purport to be complete and are qualified in their entirety by the complete agreements, which are attached as exhibits to this Annual Report on Form 20-F.
Kwang Dong Agreements
On December 22, 2008, we entered into a license agreement with Kwang Dong Pharmaceutical Co. Ltd, a South Korean limited company, or KD, and the Kwang Dong License Agreement, respectively, for the use, development and marketing of Piclidenoson in the Republic of Korea with respect to RA. In addition, the Kwang Dong License Agreement grants to KD an exclusive, royalty-free license to use certain of our trademarks, as determined from time to time, in connection with the distribution, marketing, promotion and sale of any products derived from Piclidenoson pursuant to the Kwang Dong License Agreement.
The Kwang Dong License Agreement also provides for the creation of a four member joint committee consisting of two members from each party for the purpose of serving as a joint source of experience and knowledge in Piclidenoson development and to facilitate communication and coordination between the parties with respect to such development. The joint committee will, among other things specifically identified in the Kwang Dong License Agreement, provide to the parties opinions, proposals, ideas and updates with respect to the Piclidenoson development processes conducted separately by each party.
According to the Kwang Dong License Agreement, we are entitled to receive or have received the following payments: (i) a non-refundable amount of $300,000 paid within 30 days of the effective date of the agreement; (ii) an amount of up to $1.2 million based on our compliance with certain milestones, including but not limited to, the conclusion of the Phase II clinical trial for Piclidenoson for treating RA and the receipt of various regulatory authorizations; and (iii) annual royalties of 7% of annual net sales of the licensed drug in the Republic of Korea. In addition to the amounts detailed above, we will be entitled to additional payments based on sales of raw materials to KD for the purpose of developing, producing and marketing Piclidenoson.
The Kwang Dong License Agreement is effective until KD completes all payments required thereunder, unless it is earlier terminated as a result of a material breach not cured within the specified time frame, the breach by KD of the Kwang Dong Purchase Agreement or the initiation of bankruptcy or insolvency related proceedings.
Pursuant to a share purchase agreement entered into with KD at the same time as the Kwang Dong License Agreement, KD purchased 95,304 of our ordinary shares, representing approximately 1.0 % of our share capital on a fully diluted basis, as of the date of the purchase. The shares were purchased for a premium of 50% on the shares’ average closing price for the ten days preceding December 11, 2008, or a purchase price of NIS 0.455 per share.
After the TASE approved such shares for the listing for trade on January 5, 2009, the shares were allocated to KD and the transaction was finalized in January 2009. As of December 31, 2016, KD had paid us approximately $0.8 million, which represents milestone payments pursuant to the Kwang Dong License Agreement, an advance of certain amounts to become due under the Kwang Dong License Agreement and the purchase price for the shares.
Cipher Pharmaceuticals Agreement
On March 20, 2015, we entered into a Distribution and Supply Agreement with Cipher Pharmaceuticals, or Cipher, granting Cipher the exclusive right to distribute Piclidenoson in Canada for the treatment of psoriasis and RA.
Under the Distribution and Supply Agreement, we are entitled to CDN$1.65 million upon execution of the agreement plus milestone payments upon receipt of regulatory approval by Health Canada for Piclidenoson and the first delivery of commercial launch quantities as follows (i) CDN$1 million upon the first approved indication for either psoriasis or RA, and (ii) CDN $1 million upon the second approved indication for either psoriasis or RA. In addition, following regulatory approval, we shall be entitled to a royalty of 16.5% of net sales of Piclidenoson in Canada and reimbursement for the cost of manufacturing Piclidenoson. We are also entitled to a royalty payment for any authorized generic of Piclidenoson that Cipher distributes in Canada.
| 46 |
We are responsible for supplying Cipher with finished product for distribution and conducting product development activities while Cipher is responsible for distribution, marketing and obtaining applicable regulatory approvals in Canada. The Distribution and Supply Agreement has an initial term of fifteen years, automatically renewable for additional five year periods and may be terminated in certain limited circumstances including certain breaches of the agreement and failure to achieve certain minimum quantities of sales during the contract period.
The timeline to regulatory submissions to Health Canada will be determined by the completion of the remaining clinical trial program.
CKD Agreement
On October 25, 2016, we entered into an exclusive Distribution Agreement with Chong Kun Dang Pharmaceuticals, or CKD, for the exclusive right to distribute Namodenoson for the treatment of liver cancer in South Korea, upon receipt of regulatory approvals. The Distribution Agreement further provides that we will deliver finished product to CKD and grant CKD a right of first refusal to distribute Namodenoson for other indications for which we develop Namodenoson.
The Distribution Agreement provides for up to $3,000,000 in upfront and milestone payments payable as follows: (i) an upfront payment of $500,000 within 30 days of receipt of an invoice from us, and (ii) within 30 days of the occurrence of each of the following: (1) $500,000 upon receipt by CKD of a positive result from the preliminary review by the Ministry of Food and Drug Safety, or MFDS, on obtaining orphan drug designation for Namodenoson in South Korea, (2) $500,000 upon successful completion of our ongoing Phase II clinical trial for Namodenoson, (3) $1,000,000 upon the granting of marketing authorization of Namodenoson in South Korea by the MFDS, and (4) $500,000 upon registration of Namodenoson on the “reimbursement listing” in South Korea by the National Health Insurance Services in Korea. In addition, we are entitled to a royalty of 23% of net sales of Namodenoson following commercial launch in South Korea which includes the transfer price for delivering finished product to CKD.
The Distribution Agreement has an initial term of 10 years from first commercial sale and is renewable for additional 3 year periods unless either party gives notice of termination at least 6 months prior to the then current term. The Distribution Agreement may be terminated by CKD upon 30 days prior written notice if we fail to successfully complete our ongoing Phase II clinical trial for Namodenoson and we may terminate the Distribution Agreement upon 30 days prior written notice if certain commercialization milestones are not met by CKD or certain minimum quantities of sales are not made during the contract period. In addition, either party may terminate the Distribution Agreement in the event of an uncured material breach or insolvency.
Eye-Fite Agreement
In connection with the spin-off transaction described below in “Item 10. Additional Information—Material Contracts—OphthaliX Agreements”, on November 21, 2011, we entered into a license agreement, or the Eye-Fite Agreement, with Eye-Fite according to which we (i) granted Eye-Fite a sole and exclusive worldwide license for the use of CF101 solely in the field of ophthalmic diseases and patent rights which we received under the NIH Agreement, with respect to CF101 in the field of ophthalmic diseases for research, development, commercialization and marketing throughout the world and (ii) assigned to Eye-Fite our rights, title and interest in and to any and all INDs to CF101 in the ophthalmic field. As consideration for the grant of the license, we received 999 ordinary shares of Eye-Fite, in addition to the one share we already had, which resulted in us owning all of the issued and outstanding shares of Eye-Fite, all of which were transferred to OphthaliX in connection with this transaction. Under the license agreement, Eye-fite was required to assume responsibility for making payments to our licensor, the NIH, pursuant to, and for the term of, a license agreement between us and NIH for certain patent rights relating to CF101. In June 2015, our license with NIH expired and as a result Eye-fite is no longer obligated to make any payments to NIH in connection with Can-Fite’s now expired license with NIH (other than with respect to any accrued and unpaid payments to which NIH may be entitled to). Patent rights granted to Eye-fite under the license agreement by us that are not NIH patents are free of any royalties and milestone payments.
The license agreement with Eye-Fite will remain in effect until the expiration of the last of the patents licensed thereunder, unless earlier terminated by one of the parties in accordance with its terms. We may terminate the license agreement upon customary bankruptcy and insolvency events of Eye-Fite and upon Eye-Fite’s material breach of the Eye-Fite Agreement, upon 30 days’ prior written notice. Eye-Fite may terminate the license agreement upon three months’ prior written notice for any reason and upon 30 days’ prior written notice for our material breach of the license agreement
All inventions resulting from the development and commercialization of CF101 under the license agreement belong to us, whether invented solely by us, solely by Eye-Fite or by both entities. However, the license agreement also grants Eye-Fite an exclusive license to use any such inventions in the field of ophthalmic diseases around the world for no additional consideration. Pursuant to the license agreement, we have the sole right to make elections with respect to patent term extension of or supplemental protection certificates with respect to our licensed patents and the sole right to seek and maintain any data exclusivity periods available for CF101. Also pursuant to the license agreement, we have retained the right to prosecute and maintain the patents licensed to us.
SKK Agreement
On August 27, 2015, we entered into an agreement with Japan-based Seikagaku Corporation, or SKK, terminating its license agreement with us. SKK informed us that it is strategically focused on expanding its core research and development activities in the field of glyco-science. Under the license agreement, SKK was granted a license for the use, development and marketing of Piclidenoson in Japan with respect to inflammatory indications, except for ophthalmic disease indications. The termination agreement provides, among other things, that all licenses and rights granted to SKK terminate and all clinical and non-clinical studies conducted by SKK shall be transferred free of charge to us. Over the life of the license, we received an aggregate of approximately $8.5 million from SKK.
| 47 |
Total Revenues by Category of Activity and Geographic Markets
Historically, we have generated revenues from payments received pursuant to our out-licensing agreements with Cipher, SKK and KD with respect to Piclidenoson and CKD with respect to Namodenoson. See “Item 4—Information on the Company—Business Overview—Out-Licensing and Distribution Agreements”. We recorded revenues of NIS 0.1 million for the year ended December 31, 2016 under the distribution agreement with CKD which was due to the recognition of a portion of the NIS 1.9 million ($0.5 million) advance payment received in December 2016 under the distribution agreement with CKD. We recorded revenues of NIS 0.64 million for the year ended December 31, 2015 and NIS 0.55 million for the year ended December 31, 2016 which was due to the recognition of a portion of the NIS 5.14 million (CAD 1.65 million) advance payment received in March 2015 under the distribution agreement with Cipher. In the year ended December 31, 2014 we did not record any revenues. We expect to generate future revenues through our current and potential future out-licensing arrangements with respect to Piclidenoson and Namodenoson based on the progress we make in our clinical trials.
Seasonality
Our business and operations are generally not affected by seasonal fluctuations or factors.
Raw Materials and Suppliers
We believe that the raw materials that we require to manufacture Piclidenoson, Namodenoson and CF602 are widely available from numerous suppliers and are generally considered to be generic industrial chemical supplies. We do not rely on a single or unique supplier for the current production of any therapeutic small molecule in our pipeline.
Manufacturing
We are currently manufacturing our active pharmaceutical ingredient, or API, through a leading Chinese contract research organization, or CRO. The relevant suppliers of our drug products are compliant with both current Good Manufacturing Practices, or cGMP, and current Good Laboratory Practices, or cGLP, and allow us to manufacture drug products for our current clinical trials. We anticipate that we will continue to rely on third parties to produce our drug products for clinical trials and commercialization.
There can be no assurance that our drug candidates, if approved, can be manufactured in sufficient commercial quantities, in compliance with regulatory requirements and at an acceptable cost. We and our contract manufacturers are, and will be, subject to extensive governmental regulation in connection with the manufacture of any pharmaceutical products or medical devices. We and our contract manufacturers must ensure that all of the processes, methods and equipment are compliant with cGMP for drugs on an ongoing basis, as mandated by the FDA and other regulatory authorities, and conduct extensive audits of vendors, contract laboratories and suppliers.
Contract Research Organizations
We outsource certain preclinical and clinical development activities to CROs, which in pre-clinical studies work according to cGMP and cGLP. We believe our clinical CROs comply with guidelines from the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, which attempt to harmonize the FDA and the European Medicines Agency, or the EMA, regulations and guidelines. We create and implement the drug development plans and, during the preclinical and clinical phases of development, manage the CROs according to the specific requirements of the drug candidate under development.
Marketing and Sales
We do not currently have any marketing or sales capabilities. We intend to license to, or enter into strategic alliances with, larger companies in the pharmaceutical business, which are equipped to market and/or sell our products, if any, through their well-developed marketing capabilities and distribution networks. We intend to out-license some or all of our worldwide patent rights to more than one party to achieve the fullest development, marketing and distribution of any products we develop.
| 48 |
Intellectual Property
Our success depends in part on our ability to obtain and maintain proprietary protection for our product candidates, technology and know-how, to operate without infringing the proprietary rights of others and to prevent others from infringing our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing U.S. and foreign patent applications related to our proprietary technology, inventions and improvements that we believe are important to the development of our business. We also rely on trade secrets, know-how and continuing technological innovation to develop and maintain our proprietary position.
Patents
As of March 14, 2017, we owned or exclusively licensed (from Leiden University) 12 patent families that, collectively, contain approximately 165 issued patents and pending patent applications in various countries around the world relating to our two clinical candidates, Piclidenoson and Namodenoson, and our preclinical candidate, CF602. Patents related to our drug candidates may provide future competitive advantages by providing exclusivity related to the composition of matter, formulation and method of administration of the applicable compounds and could materially improve their value. The patent positions for our leading drug candidates are described below.
With respect to our product candidates, we currently own patents and/or have patent applications pending in several countries around the world for the following families of patents:
| ● | a family of patents which pertains to the use of substances that bind to the A3AR, including Piclidenoson and Namodenoson; the pharmaceutical uses to which such family relates include the treatment of proliferative diseases, such as cancer, psoriasis and autoimmune diseases. Such patents were granted in the United States, Europe (by the European Patent Office, or the EPO, and validated in Austria, Belgium, Denmark, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Luxembourg, Portugal, Spain, Sweden, Switzerland, Holland and the United Kingdom), Australia, Canada, Israel, China, Japan, South Korea, Mexico, Poland, Russia and Hong-Kong. These patents are set to expire in 2020, other than the United States patent that will expire in 2022; |
| ● | a family of patents and a patent application which pertain to use of substances that bind to the A3AR for the treatment of viral diseases, such as AIDS and hepatitis, and which inhibit viral replication. Such patents were granted in the United States, in Europe (by the EPO and validated in France, Germany, Italy, Switzerland and the United Kingdom), Australia, China, Israel, Japan, Singapore, Canada and Hong Kong. The patent application is pending in Brazil. These patents and patent application have a filing date of January 1, 2002 and a priority date of January 16, 2001 and are set to expire in 2022, other than the United States patent that will expire in 2023; |
| ● | a patent which pertains to the use of A3AR agonists for the treatment of inflammatory arthritis, in particular RA. This patent was granted in the United States and is set to expire in 2023; |
| ● | a family of patents and patent applications which pertain to a method of identifying inflammation, determining its severity, and determining and monitoring the efficacy of the anti-inflammatory treatment by determining the level of A3AR expression in white blood cells as a biological marker for inflammation. These patents were granted in certain countries in Europe (by the EPO and validated in France, Germany, Italy, Spain, Switzerland and the United Kingdom), Australia, Israel, Japan, USA, China and Mexico. The patents are set to expire in 2025. There are patent applications pending in Canada, and Brazil. Each of the patents and patent applications has a filing date of November 30, 2005 and a priority date of December 2, 2004; |
| 49 |
| ● | a family of patents and patent applications which pertains to the use of a specific dose level of Piclidenoson (total daily dose of 4.0 mg) for the treatment of psoriasis. Such a patent was granted in Israel, Japan, United States and Europe (by the EPO and validated in in Austria, Belgium, Denmark, France, Germany, Italy, Netherlands, Spain, Sweden, Switzerland and United Kingdom). The patent is set to expire in 2030.The patent applications are pending in the China, Hong Kong, India, and South Korea, each with a filing date of September 6, 2010 and a priority date of September 6, 2009; |
| ● | a family of patents and patent applications which pertain to the method for producing Piclidenoson. Such patents were granted in U.S., India, China, Japan and Israel. These patents are set to expire in 2028. The patent applications are pending in the EPO (recently allowed, this European application designates all EPC member states) and India. Each patent and patent application has a filing date of March 13, 2008 and a priority date of March 14, 2007; |
| ● | a family of patents and patent applications which pertain to the use of A3AR agonists for the treatment of OA. Such patents were granted in Europe (by the EPO and validated in Austria, Belgium, Denmark, France, Germany, Italy, Spain, Sweden, Switzerland, Netherlands and the United Kingdom), Australia, Canada, South Korea, China, Israel, Japan and Mexico. The patents are set to expire in 2026. Patent applications are pending in the United States and Brazil. These patents and patent applications have a filing date of November 29, 2006 and a priority date of November 30, 2005; |
| ● | a family of patents and patent applications which pertains to the use of A3AR agonists for increasing liver cell division, intended to induce liver regeneration following injury or surgery. Such patents were granted in China, Israel, Japan, USA and Europe (by the EPO and validated in Austria, Belgium, Denmark, France, Germany, Italy, Netherlands, Poland, Spain, Sweden, Switzerland, United Kingdom and Turkey). There is one patent application pending in the United States which was recently allowed. Each patent or patent application in this family has a filing date of October 22, 2007 and a priority date of October 15, 2007. | |
| ● | a family of patent applications which pertain to the use of A3AR agonists for the maintenance of liver function in patients having chronic liver disease. These patent applications are pending in China, Japan, Hong-Kong. These patent applications have a filing date of January 23, 2013 and a priority date of January 23, 2012; |
| ● | a family of patent applications which pertain to treatment of sexual dysfunction. This family includes patent applications in Israel, Australia, China, Japan, Russia, Brazil, Canada, Europe, India, Mexico, South Korea, and USA. The patent applications have a filing date of August 8, 2013 with priority dates of August 8, 2012 and November 12, 2012; | |
| ● | a family of patent applications which pertain to the use of A3AR ligands for treatment of ectopic fat accumulation. This family includes a patent application in Israel and an International patent application (PCT), claiming priority from this Israeli application. The PCT application has a filing date of November 22, 2016. |
We currently hold an exclusive license from Leiden University of the Netherlands to a family of patents and patent applications that relate to the allosteric modulators of the A3AR, which includes the allosteric modulator CF602. This exclusive license relates to patents that were granted in the United States, China, Japan, South Korea and in Europe (validated in, Austria, Belgium, Denmark, France, Germany, Italy, Spain, Sweden, Switzerland, Holland and United Kingdom). There is a patent application pending in India. These granted patents and the patents that may be granted on patent applications of this patent family are set to expire in 2027.
We believe that our owned and licensed patents provide broad and comprehensive coverage of our technology, and we intend to aggressively enforce our intellectual property rights if necessary to preserve such rights and to gain the benefit of our investment. However, as a result of the termination of the NIH license agreement between Can-Fite and the NIH in June 2015 due to patent expiration, we no longer hold rights to a family of composition of matter patents relating to Piclidenoson that were licensed from NIH. Nevertheless, because Piclidenoson may be a new chemical entity, or NCE, following approval of an NDA, we, if we are the first applicant to obtain NDA approval, may be entitled to five years of data exclusivity in the United States with respect to such NCEs. Analogous data and market exclusivity provisions, of varying duration, may be available in Europe and other foreign jurisdictions. We may also be entitled to the rights under Can-Fite’s pharmaceutical use issued patents with respect to Piclidenoson, which provide patent exclusivity within the ophthalmic field until the mid-2020s. While we believe that we may be able to protect our exclusivity in the ophthalmic field through such use patent portfolio and such period of exclusivity, the lack of composition of matter patent protection may diminish our ability to maintain a proprietary position for our intended uses of Piclidenoson. Moreover, we cannot be certain that we will be the first applicant to obtain an FDA approval for any indication of Piclidenoson and we cannot be certain that we will be entitled to NCE exclusivity. In addition, we have discontinued the prosecution of a family of pending patent applications under joint ownership of Can-Fite and NIH pertaining to the use of A3AR agonists for the treatment of uveitis. Such diminution of our proprietary position could have a material adverse effect on our business, results of operation and financial condition.
| 50 |
The patent positions of companies like ours are generally uncertain and involve complex legal and factual questions. Our ability to maintain and solidify our proprietary position for our technology will depend on our success in obtaining effective claims and enforcing those claims once granted. We do not know whether any of our patent applications or those patent applications that we license will result in the issuance of any patents. Our issued patents and those that may issue in the future, or those licensed to us, may be challenged, narrowed, circumvented or found to be invalid or unenforceable, which could limit our ability to stop competitors from marketing related products or the length of term of patent protection that we may have for our products. Neither we nor our licensors can be certain that we were the first to invent the inventions claimed in our owned or licensed patents or patent applications. In addition, our competitors may independently develop similar technologies or duplicate any technology developed by us, and the rights granted under any issued patents may not provide us with any meaningful competitive advantages against these competitors. Furthermore, because of the extensive time required for development, testing and regulatory review of a potential product, before any of our products can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
Trade Secrets
We may rely, in some circumstances, on trade secrets to protect our technology. However, trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements and assignment of inventions agreements with our employees, consultants, scientific advisors and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, such agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors or others.
Scientific Advisory Board
We seek advice from our Scientific Advisory Board on scientific and medical matters generally. We call for Scientific Advisory Board meetings on an as-needed basis. The following table sets forth certain information with respect to our Scientific Advisory Board members.
| Name | Position/Institutional Affiliation | |
| Nabil Hanna, Ph.D. | Former Chief Science Officer of Biogen-Idec | |
| Kamel Khalili, Ph.D. | Temple University, Philadelphia, Pennsylvania |
| 51 |
Clinical Advisory Board
Our Clinical Advisory Board, which consists of three members, a leading U.S.-based rheumatologist, oncologist and dermatologist, plays an active role in consulting with us with respect to clinical drug development. We call for Clinical Advisory Board meetings on an as-needed basis. The following table sets forth certain information with respect to our Clinical Advisory Board members.
| Name | Position/Institutional Affiliation | |
| Dr. Michael Weinblatt | Head, Division of Rheumatology, Immunology and Allergy, Brigham and Women’s Hospital | |
| Dr. Keith Stuart | Chairman, Department of Hematology and Oncology; Professor of Medicine, Tufts University School of Medicine; Lahey Clinic Medical Center | |
| Dr. Jonathan Wilkin | Former Head, Dermatology Division, FDA |
Competition
The pharmaceutical industry is characterized by rapidly evolving technology, intense competition and a highly risky, costly and lengthy research and development process. Adequate protection of intellectual property, successful product development, adequate funding and retention of skilled, experienced and professional personnel are among the many factors critical to success in the pharmaceutical industry.
Our technology platform is based on the finding that the A3AR is highly expressed in pathological cells, such as various tumor cell types and inflammatory cells. We believe that targeting the A3AR with synthetic and highly selective A3AR agonists, such as Piclidenoson and Namodenoson, and allosteric modulators, such as CF602, induces anti-cancer and anti-inflammatory effects. Currently, our drug candidates, Piclidenoson, Namodenoson and CF602 are being developed to treat several autoimmune-inflammatory, oncological and ophthalmic indications, including but not limited to psoriasis, RA, OA, HCC, NASH. Preclinical studies have also indicated that our drug candidates have the potential to treat additional inflammatory diseases, such as sexual dysfunction, Crohn’s disease, oncological diseases and viral disease, such as the JC virus.
Despite the competition, however, we believe that our drug candidates have unique characteristics and advantages over certain drugs currently available on the market and under development to treat these indications. We believe that our pipeline of drug candidates has exhibited a potential for therapeutic success with respect to the treatment of autoimmune-inflammatory, oncological and ophthalmic diseases. We believe that targeting the A3AR with synthetic and highly selective A3AR agonists, such as Piclidenoson and Namodenoson, and allosteric modulators, such as CF602, induces anti-cancer and anti-inflammatory effects.
We believe the characteristics of Piclidenoson, as exhibited in our clinical studies to date, including its good safety profile, clinical activity, simple and less frequent delivery through oral administration and its low cost of production, position it well against the competition in the autoimmune-inflammatory markets, including the psoriasis and RA markets, where treatments, when available, often include injectable drugs, many of which can be highly toxic, expensive and not always effective. For example, while TNF inhibitor therapies transformed the treatment for many patients, a substantial percentage of patients (40% to 60%) do not respond to either disease modifying anti-rheumatic drug, or a DMARD, or biologic therapies (Simsek, 2010).
Pre-clinical pharmacology studies in different experimental animal models of arthritis revealed that Piclidenoson acts as a DMARD, which, when coupled with its good safety profile, make it competitive in the psoriasis, RA and OA markets. Our recent findings also demonstrate that a biological predictive marker can be utilized prior to treatment with Piclidenoson, which may allow it to be used as a personalized medicine therapeutic approach for the treatment of RA, potentially leading to an improvement in response rate for patients. Like Piclidenoson, Namodenoson has a good safety profile, is orally administered and has a low cost of production, which we believe positions it well in the HCC market, where only one drug, Nexavar (sorafenib), has been approved by the FDA.
In addition, our human clinical data suggests that A3AR may be a biological marker in that high A3AR expression prior to treatment has been predictive of good patient response to our drug treatment. In fact, as a result of our research we have developed a simple blood assay to test for A3AR expression as a predictive biological marker. We hold a patent with respect to the intellectual property related to such assay and are currently utilizing this assay in our ongoing Phase IIb study of Piclidenoson for the treatment of RA.
| 52 |
On the other hand, other drugs on the market, new drugs under development (including drugs that are in more advanced stages of development in comparison to our drug pipeline) and additional drugs that were originally intended for other purposes, but were found effective for purposes targeted by us, may all be competitive to the current drug candidates in our pipeline. In fact, some of these drugs are well established and accepted among patients and physicians in their respective markets, are orally bioavailable, can be efficiently produced and marketed, and are relatively safe. Moreover, other companies of various sizes engage in activities similar to ours. Most, if not all, of our competitors have substantially greater financial and other resources available to them. Competitors include companies with marketed products and/or an advanced research and development pipeline. The major competitors in the arthritis and psoriasis therapeutic field include Amgen, Centocor, Pfizer, Novartis, Abbvie, Celgene, Eli Lilly, Janssen and more. Competitors in the hepatocellular carcinoma, also known as primary liver cancer, or HCC field include companies such as Bayer. Competitors in the NASH field include companies such as Gilead, Genfit, Regato, Galmed, Allergan and Intercept. Competitors in the erectile dysfunction field include Pfizer, Eli Lilly and Bayer.
Moreover, several companies have reported the commencement of research projects related to the A3AR. Such companies include CV Therapeutics Inc. (which was acquired by Gilead), King Pharmaceuticals R&D Inv. (which was acquired by Merck), Hoechst Marion Roussel Inc., Novo Nordisk A/S and Inotek Pharmaceuticals. However, to the best of our knowledge, there is no approved drug currently on the market which is similar to our A3AR agonists, nor are we aware of any allosteric modulatorin the A3AR product pipeline similar to our allosteric modulator with respect to chemical profile and mechanism of action.
Piclidenoson for the Treatment of Psoriasis
Psoriasis is a skin condition that affects 2% to 3% of the general population according to the National Psoriasis Foundation. The disease is manifested by scaly plaques on the skin and in the severe form has a major effect on the physical and emotional well-being of the patients. Topical agents are typically used for mild disease, phototherapy for moderate disease, and systemic agents for severe disease. For moderate to severe cases, systemic biologic drugs, delivered via IV, have dominated the market. According to the National Psoriasis Foundation, common side effects of biologics include respiratory infections, flu-like symptoms, and injection site reactions while rare side effects include serious nervous system disorders, such as multiple sclerosis, seizures, or inflammation of the nerves of the eyes, blood disorders, and certain types of cancer. We believe a significant need remains for novel oral and safe drugs for patients who do not respond to existing therapies or for whom these therapies are unsuitable.
The psoriasis therapeutic market is dominated by biological drugs that are primarily administered via intravenous injection (IV) and have potential side effects. Recently, a new oral small molecule inhibitor of phosphodiesterase 4 (PDE4), Celgene’s Otezla, has gained sizable market share as a result in part due to its convenience of oral dose and comparable efficacy to the biologic drugs. In March 2016, the FDA approved Taltz (ixekizumab) by Eli Lilly. The psoriasis drug market is forecast to grow to $8.9 billion by 2018, according to estimates of Visiongain.
The current common treatments for psoriasis include topical and systemic drugs, steroids, immunosuppressive drugs such as Cyclosporine A by Novartis, MTX and biological drugs. Biological drugs, such as Enbrel (etanercept) by Amgen and Pfizer, Remicade (infliximab) by Centocor, Humira (adalimumab) by Abbvie, Stelara (ustekinumab) by Janssen, Otezla (aprelimast) by Celgene, Cosentyx (secukinumab) by Novartis and Taltz (ixekizumab) by Eli Lilly have significant side effects, are expensive and patients are often not responsive. For example, some of these drugs have received an FDA “black box” warning for increased risk of cancer in children and adolescents and risk of infection with Legionella and Listeria bacteria.
Many of the current RA drugs on the market or in development are also used for the treatment of psoriasis. See “—Piclidenoson for the Treatment of RA.” In addition, several therapies are in advanced clinical development for psoriasis and many others are in Phase II or earlier stages of development.
Piclidenoson for the Treatment of RA
RA is a severe disease that attacks approximately 0.6% of the U.S. population, mainly women and, in particular, postmenopausal women. According to Visiongain, the world RA market size is predicted to generate revenues of $34.6 billion in 2020.
| 53 |
Many drugs are used to treat RA, including DMARDs. These include MTX, plaquenil, sulfasalazine and leflunomide, all of which are small molecule drugs with mild effectiveness. MTX is the most commonly administered DMARD for RA. It is a generic chemotherapeutic agent marketed by several manufacturers that is administered orally. Due to its relatively toxic nature, however, MTX may result in severe side effects including sores, anemia, diarrhea, nausea/vomiting, abdominal pain, bruising/bleeding, and liver problems.
The second class of DMARD includes biological drugs, such as Enbrel (etanercept) by Amgen, Remicade (infliximab) by Centocor, and Humira (adalimumab) by Abbvie. These drugs are usually administered in combination with MTX and are more effective in combination, but may have severe side effects, including risk of lymphoma and serious infection. Biological drugs are administered through injection, are generally expensive and there is no biomarker to predict the response, if any. As such, response rates typically range between 40-60% (Simsek, 2010). Steroidal drugs are also used to reduce the general activity of the immune system and for pain relief. In addition, the FDA recently approved Pfizer’s Xeljanz (tofacitinib) small molecule drug, which is the first JAK inhibitor drug, or a drug that inhibits the effect of one or more of the enzymes in the janus kinase family, or a family enzymes that transfer cytokine-mediated signals, to treat RA. Moreover, several therapies, including biological drugs and small molecule drugs, are in advanced clinical development for RA including baricitninib by Eli Lilly which is pending FDA approval, while others are in Phase II or earlier stages of development.
Namodenoson for the Treatment of HCC
According to the Living with Liver Cancer HCC is the sixth most common form of cancer, the most common form of liver cancer in adults and the third most common cause of cancer-related mortality worldwide, particularly in Asia. According to the American Cancer Society, more than 700,000 people are diagnosed with liver cancer each year throughout the world and more than 600,000 persons die from liver cancer each year. Nexavar (sorafenib) by Bayer is the only approved drug for HCC and prolongs patient survival time by only a few months. According to Datamonitor, the HCC drug market is expected to reach $1.4 billion by 2019.
Several therapies are in advanced clinical development for HCC including Opdivo (nivolumab) by Bristol-Myers and Cabozantinib by Exelixis. Some drugs under development act as a single agent and some act in combination with Nexavar. Moreover, some are first line treatments while others are second line treatments. In addition, many existing approaches are used in the treatment of unresectable liver cancer, including alcohol injection, radiofrequency ablation, chemoembolization, cryoablation and radiation therapy.
Namodenoson for the Treatment of NASH
Rates of NAFLD and NASH are increasing in the U.S. in concert with increasing rates of obesity and diabetes. In fact, NASH is now the third leading cause of liver transplant in the U.S. It is estimated that 17-33% of Americans have fatty liver, with approximately one-third going on to develop NASH. NASH is believed to affect 2-5% of adult Americans. Despite the progression of several interesting clinical-stage candidates by companies such as Gilead, Genfit, Regado, Conatus, Galmed, Allergan and Intercept as well as others, there are currently no FDA approved treatment options for NASH.
By 2025, Deutsche Bank estimates the addressable pharmaceutical market for NASH will reach $35-40 billion in size.
CF602 for the Treatment of Erectile Dysfunction
According to a the Massachusetts Male Aging Study in 1994, 52% of the respondents between the ages of 40 and 70 years old reported some degree of erectile dysfunction.
The most popular class of drug to treat erectile dysfunction is the phosophodiesterase type 5 inhibitors, or PDE5. These drugs block the degradative action of cyclic guanosine monophosphate, or GMP, specific phosphodiesterase type 5 on cyclic GMP in the smooth muscle cells lining the blood vessels supplying the corpus cavernosum of the penis. An erection is caused by increased blood flow into the penis resulting from the relaxation of penile arteries and corpus cavernosal smooth muscle. This response is mediated by the release of nitric oxide from nerve terminals and endothelial cells, which stimulates the synthesis of cyclic GMP in smooth muscle cells. The inhibition of PDE5 enhances erectile function by increasing the concentration of cyclic GMP in the corpus cavernosum and pulmonary arteries.
Unfortunately, the systemic side effects of PDE5 inhibitors include a decrease in sitting blood pressure. This has resulted in warnings and precautions and contraindications of use in patients already taking antihypertensive agents like nitrates or alpha-blockers. A study published in the American Journal of Medicine (Selvin E., et al., 2007) found that persons with a history of heart disease, hypertension, and diabetes had a higher probability of impotence. A second study published in the same journal (Shah NP., et al, 2015) notes that vascular erectile dysfunction is a powerful marker of increased cardiovascular risk. We believe a significant market opportunity exists targeting erectile dysfunction patients contraindicated for use of the market leading products, Viagra and Cialis.
GlobalData estimates the value of the erectile dysfunction therapeutic market to be approximately $2.6 billion by 2018 with few drugs on the market which includes Viagra (sildenafil) by Pfizer, Cialis (tadalafil) by Eli Lilly and Levitra (vardenafil) by Bayer.
| 54 |
Insurance
We maintain insurance for our offices and laboratory in Petah-Tikva, Israel. Our insurance program covers approximately $0.375 million of equipment and lease improvements against risk of loss, excluding damage from inventory theft. In addition, we maintain the following insurance: employer liability with coverage of approximately $5.0 million; third party liability with coverage of approximately $0.75 million; fire insurance coverage of approximately $0.725 million; natural disaster coverage of approximately $1.1 million; all risk coverage of approximately $0.02 million for electronic equipment and machinery insurance for laboratory refrigerators; and directors’ and officers’ liability with coverage of $2.0 million per claim and $10.0 million in the aggregate.
We also maintain worldwide product and clinical trial liability insurance with coverage of approximately $5 million with respect to the Piclidenoson and Namodenoson drugs used in clinical trials. We also procure additional insurance for each specific clinical trial which covers a certain number of trial participants and which varies based on the particular clinical trial. Certain of such policies are based on the Declaration of Helsinki, which is a set of ethical principles regarding human experimentation developed for the medical community by the World Medical Association, and certain protocols of the Israeli Ministry of Health.
We procure cargo marine coverage when we ship substances for our clinical studies. Such insurance is custom-fit to the special requirements of the applicable shipment, such as temperature and/or climate sensitivity. If required, we insure the substances to the extent they are stored in central depots and at clinical sites.
We believe that our insurance policies are adequate and customary for a business of our kind. However, because of the nature of our business, we cannot assure you that we will be able to maintain insurance on a commercially reasonable basis or at all, or that any future claims will not exceed our insurance coverage.
Environmental Matters
We are subject to various environmental, health and safety laws and regulations, including those governing air emissions, water and wastewater discharges, noise emissions, the use, management and disposal of hazardous, radioactive and biological materials and wastes and the cleanup of contaminated sites. We believe that our business, operations and facilities are being operated in compliance in all material respects with applicable environmental and health and safety laws and regulations. Our laboratory personnel in Israel have ongoing communication with the Israeli Ministry of Environmental Protection in order to verify compliance with relevant instructions and regulations. In addition, all of our laboratory personnel participate in instruction on the proper handling of chemicals, including hazardous substances before commencing employment, and during the course of their employment, with us. In addition, all information with respect to any chemical substance that we use is filed and stored as a Material Safety Data Sheet, as required by applicable environmental regulations. Based on information currently available to us, we do not expect environmental costs and contingencies to have a material adverse effect on us. The operation of our testing facilities, however, entails risks in these areas. Significant expenditures could be required in the future if these facilities are required to comply with new or more stringent environmental or health and safety laws, regulations or requirements. See “Business — Government Regulation and Funding — Israel Ministry of Environment — Toxin Permit.”
Government Regulation and Funding
We operate in a highly controlled regulatory environment. Stringent regulations establish requirements relating to analytical, toxicological and clinical standards and protocols in respect of the testing of pharmaceuticals. Regulations also cover research, development, manufacturing and reporting procedures, both pre- and post-approval. In many markets, especially in Europe, marketing and pricing strategies are subject to national legislation or administrative practices that include requirements to demonstrate not only the quality, safety and efficacy of a new product, but also its cost-effectiveness relating to other treatment options. Failure to comply with regulations can result in stringent sanctions, including product recalls, withdrawal of approvals, seizure of products and criminal prosecution.
| 55 |
Before obtaining regulatory approvals for the commercial sale of our product candidates, we must demonstrate through preclinical studies and clinical trials that our product candidates are safe and effective. Historically, the results from preclinical studies and early clinical trials often have not accurately predicted results of later clinical trials. In addition, a number of pharmaceutical products have shown promising results in clinical trials but subsequently failed to establish sufficient safety and efficacy results to obtain necessary regulatory approvals. We have incurred and will continue to incur substantial expense for, and devote a significant amount of time to, preclinical studies and clinical trials. Many factors can delay the commencement and rate of completion of clinical trials, including the inability to recruit patients at the expected rate, the inability to follow patients adequately after treatment, the failure to manufacture sufficient quantities of materials used for clinical trials, and the emergence of unforeseen safety issues and governmental and regulatory delays. If a product candidate fails to demonstrate safety and efficacy in clinical trials, this failure may delay development of other product candidates and hinder our ability to conduct related preclinical studies and clinical trials. Additionally, as a result of these failures, we may also be unable to obtain additional financing.
Governmental authorities in all major markets require that a new pharmaceutical product be approved or exempted from approval before it is marketed, and have established high standards for technical appraisal, which can result in an expensive and lengthy approval process. The time to obtain approval varies by country and some products are never approved. The lengthy process of conducting clinical trials, seeking approval and the subsequent compliance with applicable statutes and regulations, if approval is obtained, are very costly and require the expenditure of substantial resources.
A summary of the U.S., EU and Israeli regulatory processes follow below.
United States
In the United States, the Public Health Service Act and the Federal Food, Drug, and Cosmetic Act, as amended, and the regulations promulgated thereunder, and other federal and state statutes and regulations govern, among other things, the safety and effectiveness standards for our products and the raw materials and components used in the production of, testing, manufacture, labeling, storage, record keeping, approval, advertising and promotion of our products on a product-by-product basis.
Preclinical tests include in vitro and in vivo evaluation of the product candidate, its chemistry, formulation and stability, and animal studies to assess potential safety and efficacy. Certain preclinical tests must be conducted in compliance with good laboratory practice regulations. Violations of these regulations can, in some cases, lead to invalidation of the studies, requiring them to be replicated. After laboratory analysis and preclinical testing, testing, a sponsor files an Investigational New Drug application, or IND, to begin human testing. Typically, a manufacturer conducts a three-phase human clinical testing program which itself is subject to numerous laws and regulatory requirements, including adequate monitoring, reporting, record keeping and informed consent. In Phase I, small clinical trials are conducted to determine the safety and proper dose ranges of our product candidates. In Phase II, clinical trials are conducted to assess safety and gain preliminary evidence of the efficacy of our product candidates. In Phase III, clinical trials are conducted to provide sufficient data for the statistically valid evidence of safety and efficacy. The time and expense required for us to perform this clinical testing can vary and is substantial. We cannot be certain that we will successfully complete Phase I, Phase II or Phase III testing of our product candidates within any specific time period, if at all. Furthermore, the FDA, the Institutional Review Board responsible for approving and monitoring the clinical trials at a given site, the Data Safety Monitoring Board, where one is used, or we may suspend the clinical trials at any time on various grounds, including a finding that subjects or patients are exposed to unacceptable health risk.
If the clinical data from these clinical trials (Phases I, II and III) are deemed to support the safety and effectiveness of the candidate product for its intended use, then we may proceed to seek to file with the FDA, a New Drug Application, or NDA, seeking approval to market a new drug for one or more specified intended uses. We have not completed our clinical trials for any candidate product for any intended use and therefore, we cannot ascertain whether the clinical data will support and justify filing an NDA. Nevertheless, if and when we are able to ascertain that the clinical data supports and justifies filing an NDA, we intend to make such appropriate filings.
The purpose of the NDA is to provide the FDA with sufficient information so that it can assess whether it ought to approve the candidate product for marketing for specific intended uses. The fact that the FDA has designated a drug as an orphan drug for a particular intended use does not mean that the drug has been approved for marketing. Only after an NDA has been approved by the FDA is marketing appropriate. A request for orphan drug status must be filed before the NDA is filed. The orphan drug designation, though, provides certain benefits, including a seven-year period of market exclusivity subject to certain exceptions. In February 2012, the FDA granted an orphan drug status for the active moiety, or the part of the drug that is responsible for the physiological or pharmacological action of the drug substance, of Namodenoson for the treatment of HCC. Subsequently, in October 2015, the EMA granted Namodenoson orphan drug designation for the treatment of HCC. See “Item 4. Information on the Company—B. Business Overview—Namodenoson”.
| 56 |
The NDA normally contains, among other things, sections describing the chemistry, manufacturing, and controls, non-clinical pharmacology and toxicology, human pharmacokinetics and bioavailability, microbiology, the results of the clinical trials, and the proposed labeling which contains, among other things, the intended uses of the candidate product.
We cannot take any action to market any new drug or biologic product in the United States until our appropriate marketing application has been approved by the FDA. The FDA has substantial discretion over the approval process and may disagree with our interpretation of the data submitted. The process may be significantly extended by requests for additional information or clarification regarding information already provided. As part of this review, the FDA may refer the application to an appropriate advisory committee, typically a panel of clinicians. Satisfaction of these and other regulatory requirements typically takes several years, and the actual time required may vary substantially based upon the type, complexity and novelty of the product. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures on our activities. We cannot be certain that the FDA or other regulatory agencies will approve any of our products on a timely basis, if at all. Success in preclinical or early stage clinical trials does not assure success in later-stage clinical trials. Even if a product receives regulatory approval, the approval may be significantly limited to specific indications or uses and these limitations may adversely affect the commercial viability of the product. Delays in obtaining, or failures to obtain regulatory approvals, would have a material adverse effect on our business.
Even after we obtain FDA approval, we may be required to conduct further clinical trials (i.e., Phase IV trials) and provide additional data on safety and effectiveness. We are also required to gain separate approval for the use of an approved product as a treatment for indications other than those initially approved. In addition, side effects or adverse events that are reported during clinical trials can delay, impede or prevent marketing approval. Similarly, adverse events that are reported after marketing approval can result in additional limitations being placed on the product’s use and, potentially, withdrawal of the product from the market. Any adverse event, either before or after marketing approval, can result in product liability claims against us.
As an alternate path for FDA approval of new indications or new formulations of previously-approved products, a company may file a Section 505(b)(2) NDA, instead of a “stand-alone” or “full” NDA. Section 505(b)(2) of the Food, Drug, and Cosmetic Act, or FDC, was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Amendments. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. Some examples of products that may be allowed to follow a 505(b)(2) path to approval are drugs that have a new dosage form, strength, route of administration, formulation or indication. The Hatch-Waxman Amendments permit the applicant to rely upon certain published nonclinical or clinical studies conducted for an approved product or the FDA’s conclusions from prior review of such studies. The FDA may require companies to perform additional studies or measurements to support any changes from the approved product. The FDA may then approve the new product for all or some of the labeled indications for which the reference product has been approved, as well as for any new indication supported by the NDA. While references to nonclinical and clinical data not generated by the applicant or for which the applicant does not have a right of reference are allowed, all development, process, stability, qualification and validation data related to the manufacturing and quality of the new product must be included in an NDA submitted under Section 505(b)(2).
To the extent that the Section 505(b)(2) applicant is relying on the FDA’s conclusions regarding studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book publication. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. The Section 505(b)(2) application also will not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the reference product has expired. Thus, the Section 505(b)(2) applicant may invest a significant amount of time and expense in the development of its products only to be subject to significant delay and patent litigation before its products may be commercialized.
In addition to regulating and auditing human clinical trials, the FDA regulates and inspects equipment, facilities, laboratories and processes used in the manufacturing and testing of such products prior to providing approval to market a product. If after receiving FDA approval, we make a material change in manufacturing equipment, location or process, additional regulatory review and approval may be required. We also must adhere to cGMP regulations and product-specific regulations enforced by the FDA through its facilities inspection program. The FDA also conducts regular, periodic visits to re-inspect our equipment, facilities, laboratories and processes following the initial approval. If, as a result of these inspections, the FDA determines that our equipment, facilities, laboratories or processes do not comply with applicable FDA regulations and conditions of product approval, the FDA may seek civil, criminal or administrative sanctions and/or remedies against us, including the suspension of our manufacturing operations.
| 57 |
We have currently received no approvals to market our products from the FDA or other foreign regulators.
We are also subject to various federal, state and international laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. The federal Anti-kickback law, which governs federal healthcare programs (e.g., Medicare, Medicaid), makes it illegal to solicit, offer, receive or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. Many states have similar laws that are not restricted to federal healthcare programs. Federal and state false claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment to third party payers (including Medicare and Medicaid), claims for reimbursement, including claims for the sale of drugs or services, that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. If the government or a whistleblower were to allege that we violated these laws there could be a material adverse effect on us, including our stock price. Even an unsuccessful challenge could cause adverse publicity and be costly to respond to, which could have a materially adverse effect on our business, results of operations and financial condition. A finding of liability under these laws can have significant adverse financial implications for us and can result in payment of large penalties and possible exclusion from federal healthcare programs. We will consult counsel concerning the potential application of these and other laws to our business and our sales, marketing and other activities and will make good faith efforts to comply with them. However, given their broad reach and the increasing attention given by law enforcement authorities, we cannot assure you that some of our activities will not be challenged or deemed to violate some of these laws.
European Union
Regulation and Marketing Authorization in the European Union
The process governing approval of medicinal products in the European Union follows essentially the same lines as in the United States and, likewise, generally involves satisfactorily completing each of the following:
| ● | preclinical laboratory tests, animal studies and formulation studies all performed in accordance with the applicable E.U. Good Laboratory Practice regulations; | |
| ● | submission to the relevant national authorities of a clinical trial application, or CTA, which must be approved before human clinical trials may begin; | |
| ● | performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the product for each proposed indication; | |
| ● | submission to the relevant competent authorities of a marketing authorization application, or MAA, which includes the data supporting safety and efficacy as well as detailed information on the manufacture and composition of the product in clinical development and proposed labelling; | |
| ● | satisfactory completion of an inspection by the relevant national authorities of the manufacturing facility or facilities, including those of third parties, at which the product is produced to assess compliance with strictly enforced current cGMP; | |
| ● | potential audits of the non-clinical and clinical trial sites that generated the data in support of the MAA; and | |
| ● | review and approval by the relevant competent authority of the MAA before any commercial marketing, sale or shipment of the product. |
| 58 |
Preclinical Studies
Preclinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animal studies, in order to assess the potential safety and efficacy of the product. The conduct of the preclinical tests and formulation of the compounds for testing must comply with the relevant E.U. regulations and requirements. The results of the preclinical tests, together with relevant manufacturing information and analytical data, are submitted as part of the CTA.
Clinical Trial Approval
Requirements for the conduct of clinical trials in the European Union including Good Clinical Practice, or GCP, are implemented in the Clinical Trials Directive 2001/20/EC and the GCP Directive 2005/28/EC. Pursuant to Directive 2001/20/EC and Directive 2005/28/EC, as amended, a system for the approval of clinical trials in the European Union has been implemented through national legislation of the member states. Under this system, approval must be obtained from the competent national authority of an E.U. member state in which a study is planned to be conducted, or in multiple member states if the clinical trial is to be conducted in a number of member states. To this end, a CTA is submitted, which must be supported by an investigational medicinal product dossier, or IMPD, and further supporting information prescribed by Directive 2001/20/EC and Directive 2005/28/EC and other applicable guidance documents. Furthermore, a clinical trial may only be started after a competent ethics committee has issued a favorable opinion on the clinical trial application in that country.
In April 2014, a new Clinical Trials Regulation, (EU) No 536/2014 was adopted which will replace the current Clinical Trials Directive 2001/20/EC. To ensure that the rules for clinical trials are identical throughout the European Union, the new E.U. clinical trials legislation was passed as a “regulation” that is directly applicable in all E.U. member states. All clinical trials performed in the European Union are required to be conducted in accordance with the Clinical Trials Directive 2001/20/EC until the new Clinical Trials Regulation (EU) No 536/2014 becomes applicable, which is scheduled to be in 2018.
The new Regulation (EU) No 536/2014 aims to harmonize, simplify and streamline the approval of clinical trials in the European Union. The main characteristics of the Regulation include:
| ● | A streamlined application procedure via a single entry point, the E.U. portal. | |
| ● | A single set of documents to be prepared and submitted for the application as well as simplified reporting procedures that will spare sponsors from submitting broadly identical information separately to various bodies and different member states. | |
| ● | A harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts. Part I is assessed jointly by all member states concerned. Part II is assessed separately by each member state concerned. | |
| ● | Strictly defined deadlines for the assessment of clinical trial application. | |
| ● | The involvement of the ethics committees in the assessment procedure in accordance with the national law of the member state concerned but within the overall timelines defined by the Regulation (EU) No 536/2014. |
Marketing Authorization
Authorization to market a product in the member states of the European Union proceeds under one of four procedures: a centralized authorization procedure, a mutual recognition procedure, a decentralized procedure or a national procedure.
| 59 |
Centralized Authorization Procedure
The centralized procedure enables applicants to obtain a marketing authorization that is valid in all E.U. member states based on a single application. Certain medicinal products, including products developed by means of biotechnological processes, must undergo the centralized authorization procedure for marketing authorization, which, if granted by the European Commission, is automatically valid in all 28 E.U. member states. The EMA and the European Commission administer this centralized authorization procedure pursuant to Regulation (EC) No 726/2004.
Pursuant to Regulation (EC) No 726/2004, this procedure is mandatory for:
| ● | medicinal products developed by means of one of the following biotechnological processes: | |
| ● | recombinant DNA technology; | |
| ● | controlled expression of genes coding for biologically active proteins in prokaryotes and eukaryotes including transformed mammalian cells; and | |
| ● | hybridoma and monoclonal antibody methods; | |
| ● | advanced therapy medicinal products as defined in Article 2 of Regulation (EC) No. 1394/2007 on advanced therapy medicinal products; | |
| ● | medicinal products for human use containing a new active substance that, on the date of effectiveness of this regulation, was not authorized in the European Union, and for which the therapeutic indication is the treatment of any of the following diseases: | |
| ● | acquired immune deficiency syndrome; | |
| ● | cancer; | |
| ● | neurodegenerative disorder; | |
| ● | diabetes; | |
| ● | auto-immune diseases and other immune dysfunctions; and | |
| ● | viral diseases; and | |
| ● | medicinal products that are designated as orphan medicinal products pursuant to Regulation (EC) No 141/2000. |
The centralized authorization procedure is optional for other medicinal products if they contain a new active substance or if the applicant shows that the medicinal product concerned constitutes a significant therapeutic, scientific or technical innovation or that the granting of authorization is in the interest of patients in the European Union.
Administrative Procedure
Under the centralized authorization procedure, the EMA’s Committee for Human Medicinal Products, or CHMP, serves as the scientific committee that renders opinions about the safety, efficacy and quality of medicinal products for human use on behalf of the EMA. The CHMP is composed of experts nominated by each member state’s national authority for medicinal products, with expert appointed to act as Rapporteur for the co-ordination of the evaluation with the possible assistance of a further member of the Committee acting as a Co-Rapporteur. After approval, the Rapporteur(s) continue to monitor the product throughout its life cycle. The CHMP has 210 days to adopt an opinion as to whether a marketing authorization should be granted. The process usually takes longer in case additional information is requested, which triggers clock-stops in the procedural timelines. The process is complex and involves extensive consultation with the regulatory authorities of member states and a number of experts. When an application is submitted for a marketing authorization in respect of a drug that is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation, the applicant may pursuant to Article 14(9) Regulation (EC) No 726/2004 request an accelerated assessment procedure. If the CHMP accepts such request, the time-limit of 210 days will be reduced to 150 days but it is possible that the CHMP can revert to the standard time-limit for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment. Once the procedure is completed, a European Public Assessment Report, or EPAR, is produced. If the opinion is negative, information is given as to the grounds on which this conclusion was reached. After the adoption of the CHMP opinion, a decision on the MAA must be adopted by the European Commission, after consulting the E.U. member states, which in total can take more than 60 days.
| 60 |
Conditional Approval
In specific circumstances, E.U. legislation (Article 14(7) Regulation (EC) No 726/2004 and Regulation (EC) No 507/2006 on Conditional Marketing Authorisations for Medicinal Products for Human Use) enables applicants to obtain a conditional marketing authorization prior to obtaining the comprehensive clinical data required for an application for a full marketing authorization. Such conditional approvals may be granted for product candidates (including medicines designated as orphan medicinal products) if (1) the risk-benefit balance of the product candidate is positive, (2) it is likely that the applicant will be in a position to provide the required comprehensive clinical trial data, (3) the product fulfills unmet medical needs and (4) the benefit to public health of the immediate availability on the market of the medicinal product concerned outweighs the risk inherent in the fact that additional data are still required. A conditional marketing authorization may contain specific obligations to be fulfilled by the marketing authorization holder, including obligations with respect to the completion of ongoing or new studies, and with respect to the collection of pharmacovigilance data. Conditional marketing authorizations are valid for one year, and may be renewed annually, if the risk-benefit balance remains positive, and after an assessment of the need for additional or modified conditions and/or specific obligations. The timelines for the centralized procedure described above also apply with respect to the review by the CHMP of applications for a conditional marketing authorization.
Marketing Authorization under Exceptional Circumstances
Under Article 14(8) Regulation (EC) No 726/2004, products for which the applicant can demonstrate that comprehensive data (in line with the requirements laid down in Annex I of Directive 2001/83/EC, as amended) cannot be provided (due to specific reasons foreseen in the legislation) might be eligible for marketing authorization under exceptional circumstances. This type of authorization is reviewed annually to reassess the risk-benefit balance. The fulfillment of any specific procedures/obligations imposed as part of the marketing authorization under exceptional circumstances is aimed at the provision of information on the safe and effective use of the product and will normally not lead to the completion of a full dossier/approval.
Market Authorizations Granted by Authorities of E.U. Member States
In general, if the centralized procedure is not followed, there are three alternative procedures as prescribed in Directive 2001/83/EC:
| ● | The decentralized procedure allows applicants to file identical applications to several E.U. member states and receive simultaneous national approvals based on the recognition by E.U. member states of an assessment by a reference member state. | |
| ● | The national procedure is only available for products intended to be authorized in a single E.U. member state. | |
| ● | A mutual recognition procedure similar to the decentralized procedure is available when a marketing authorization has already been obtained in at least one E.U. member state. |
A marketing authorization may be granted only to an applicant established in the European Union.
Pediatric Studies
Prior to obtaining a marketing authorization in the European Union, applicants have to demonstrate compliance with all measures included in an EMA-approved Paediatric Investigation Plan, or PIP, covering all subsets of the paediatric population, unless the EMA has granted a product-specific waiver, a class waiver, or a deferral for one or more of the measures included in the PIP. The respective requirements for all marketing authorization procedures are set forth in Regulation (EC) No 1901/2006, which is referred to as the Pediatric Regulation. This requirement also applies when a company wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already authorized. The Pediatric Committee of the EMA, or PDCO, may grant deferrals for some medicines, allowing a company to delay development of the medicine in children until there is enough information to demonstrate its effectiveness and safety in adults. The PDCO may also grant waivers when development of a medicine in children is not needed or is not appropriate, such as for diseases that only affect the elderly population.
Before a marketing authorization application can be filed, or an existing marketing authorization can be amended, the EMA determines that companies actually comply with the agreed studies and measures listed in each relevant PIP.
| 61 |
Periods of Authorization and Renewals
A marketing authorization is valid for five years in principle and the marketing authorization may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance by the competent authority of the authorizing member state. To this end, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least six months before the marketing authorization ceases to be valid. Once renewed, the marketing authorization is valid for an unlimited period, unless the European Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal. Any authorization which is not followed by the actual placing of the drug on the E.U. market (in case of centralized procedure) or on the market of the authorizing member state within three years after authorization ceases to be valid (the so-called sunset clause).
Orphan Drug Designation and Exclusivity
Pursuant to Regulation (EC) No 141/2000 and Regulation (EC) No. 847/2000, the European Commission can grant such orphan medicinal product designation to products for which the sponsor can establish that it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 people in the European Union, or a life threatening, seriously debilitating or serious and chronic condition in the European Union and that without incentives it is unlikely that sales of the drug in the European Union would generate a sufficient return to justify the necessary investment. In addition, the sponsor must establish that there is no other satisfactory method approved in the European Union of diagnosing, preventing or treating the condition, or if such a method exists, the proposed orphan drug will be of significant benefit to patients.
Orphan drug designation is not a marketing authorization. It is a designation that provides a number of benefits, including fee reductions, regulatory assistance, and the possibility to apply for a centralized E.U. marketing authorization, as well as ten years of market exclusivity following a marketing authorization. During this market exclusivity period, neither the EMA, the European Commission nor the member states can accept an application or grant a marketing authorization for a “similar medicinal product.” A “similar medicinal product” is defined as a medicinal product containing a similar active substance or substances as those contained in an authorized orphan medicinal product and that is intended for the same therapeutic indication. The market exclusivity period for the authorized therapeutic indication may be reduced to six years if, at the end of the fifth year, it is established that the orphan designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. In addition, a competing similar medicinal product may in limited circumstances be authorized prior to the expiration of the market exclusivity period, including if it is shown to be safer, more effective or otherwise clinically superior to the already approved orphan drug. Furthermore, a product can lose orphan designation, and the related benefits, prior to us obtaining a marketing authorization if it is demonstrated that the orphan designation criteria are no longer met.
Regulatory Data Protection
E.U. legislation also provides for a system of regulatory data and market exclusivity. According to Article 14(11) of Regulation (EC) No 726/2004, as amended, and Article 10(1) of Directive 2001/83/EC, as amended, upon receiving marketing authorization, new chemical entities approved on the basis of complete independent data package benefit from eight years of data exclusivity and an additional two years of market exclusivity. Data exclusivity prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic (abbreviated) application. During the additional two-year period of market exclusivity, a generic marketing authorization can be submitted, and the innovator’s data may be referenced, but no generic medicinal product can be marketed until the expiration of the market exclusivity. The overall ten-year period will be extended to a maximum of 11 years if, during the first eight years of those ten years, the marketing authorization holder, or MAH, obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. Even if a compound is considered to be a new chemical entity and the innovator is able to gain the period of data exclusivity, another company nevertheless could also market another version of the drug if such company obtained marketing authorization based on an MAA with a complete independent data package of pharmaceutical test, preclinical tests and clinical trials. However, products designated as orphan medicinal products enjoy, upon receiving marketing authorization, a period of ten years of orphan market exclusivity—see also Orphan Drug Designation and Exclusivity. Depending upon the timing and duration of the E.U. marketing authorization process, products may be eligible for up to five years’ supplementary protection certificates, or SPCs, pursuant to Regulation (EC) No 469/2009. Such SPCs extend the rights under the basic patent for the drug.
| 62 |
Regulatory Requirements After a Marketing Authorization has been Obtained
If we obtain authorization for a medicinal product in the European Union, we will be required to comply with a range of requirements applicable to the manufacturing, marketing, promotion and sale of medicinal products:
Pharmacovigilance and other requirements
We will, for example, have to comply with the E.U.’s stringent pharmacovigilance or safety reporting rules, pursuant to which post-authorization studies and additional monitoring obligations can be imposed. Other requirements relate, for example, to the manufacturing of products and APIs in accordance with good manufacturing practice standards. E.U. regulators may conduct inspections to verify our compliance with applicable requirements, and we will have to continue to expend time, money and effort to remain compliant. Non-compliance with E.U. requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties in the European Union. Similarly, failure to comply with the E.U.’s requirements regarding the protection of individual personal data can also lead to significant penalties and sanctions. Individual E.U. member states may also impose various sanctions and penalties in case we do not comply with locally applicable requirements.
Manufacturing
The manufacturing of authorized drugs, for which a separate manufacturer’s license is mandatory, must be conducted in strict compliance with the EMA’s Good Manufacturing Practices, or GMP, requirements and comparable requirements of other regulatory bodies in the European Union, which mandate the methods, facilities and controls used in manufacturing, processing and packing of drugs to assure their safety and identity. The EMA enforces its current GMP requirements through mandatory registration of facilities and inspections of those facilities. The EMA may have a coordinating role for these inspections while the responsibility for carrying them out rests with the member states competent authority under whose responsibility the manufacturer falls. Failure to comply with these requirements could interrupt supply and result in delays, unanticipated costs and lost revenues, and could subject the applicant to potential legal or regulatory action, including but not limited to warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil and criminal penalties.
Marketing and Promotion
The marketing and promotion of authorized drugs, including industry-sponsored continuing medical education and advertising directed toward the prescribers of drugs and/or the general public, are strictly regulated in the European Union under Directive 2001/83/EC. The applicable regulations aim to ensure that information provided by holders of marketing authorizations regarding their products is truthful, balanced and accurately reflects the safety and efficacy claims authorized by the EMA or by the competent authority of the authorizing member state. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties.
Patent Term Extension
In order to compensate the patentee for delays in obtaining a marketing authorization for a patented product, a supplementary certificate, or SPC, may be granted extending the exclusivity period for that specific product by up to five years. Applications for SPCs must be made to the relevant patent office in each E.U. member state and the granted certificates are valid only in the member state of grant. An application has to be made by the patent owner within six months of the first marketing authorization being granted in the European Union (assuming the patent in question has not expired, lapsed or been revoked) or within six months of the grant of the patent (if the marketing authorization is granted first). In the context of SPCs, the term “product” means the active ingredient or combination of active ingredients for a medicinal product and the term “patent” means a patent protecting such a product or a new manufacturing process or application for it. The duration of an SPC is calculated as the difference between the patent’s filing date and the date of the first marketing authorization, minus five years, subject to a maximum term of five years.
| 63 |
A six month pediatric extension of an SPC may be obtained where the patentee has carried out an agreed pediatric investigation plan, the authorized product information includes information on the results of the studies and the product is authorized in all member states of the European Union.
Israel
Israel Ministry of the Environment — Toxin Permit
In accordance with the Israeli Dangerous Substance Law — 1993, the Ministry of the Environment may grant a permit in order to use toxic materials. Because we utilize toxic materials in the course of operation of our laboratories, we were required to apply for a permit to use these materials. Our current toxin permit will remain in effect until January 2020.
Other Licenses and Approvals
We have a business license from the municipality of Petah-Tikva for a drug development research laboratory located at our offices in Petah Tikva, Israel. In order to obtain this license, we also received approval from the Petah-Tikva Association of Towns Fire Department. The business license is valid until December 2017. We also have a radioactive materials or products containing radioactive materials license, which is valid until July 2017.
In 2002, we received approval from the National Council on Animal Experiments, approving us as an institution authorized to conduct experiments on animals.
Clinical Testing in Israel
In order to conduct clinical testing on humans in Israel, special authorization must first be obtained from the ethics committee and general manager of the institution in which the clinical studies are scheduled to be conducted, as required under the Guidelines for Clinical Trials in Human Subjects implemented pursuant to the Israeli Public Health Regulations (Clinical Trials in Human Subjects), as amended from time to time, and other applicable legislation. These regulations also require authorization from the Israeli Ministry of Health, except in certain circumstances, and in the case of genetic trials, special fertility trials and similar trials, an additional authorization of the overseeing institutional ethics committee. The institutional ethics committee must, among other things, evaluate the anticipated benefits that are likely to be derived from the project to determine if it justifies the risks and inconvenience to be inflicted on the human subjects, and the committee must ensure that adequate protection exists for the rights and safety of the participants as well as the accuracy of the information gathered in the course of the clinical testing. Since we intend to perform a portion of the clinical studies on certain of our product candidates in Israel, we will be required to obtain authorization from the ethics committee and general manager of each institution in which we intend to conduct our clinical trials, and in most cases, from the Israeli Ministry of Health.
Israel Ministry of Health
Israel’s Ministry of Health, which regulates medical testing, has adopted protocols that correspond, generally, to those of the FDA and the EMA, making it comparatively straightforward for studies conducted in Israel to satisfy FDA and the European Medicines Agency requirements, thereby enabling medical technologies subjected to clinical trials in Israel to reach U.S. and EU commercial markets in an expedited fashion. Many members of Israel’s medical community have earned international prestige in their chosen fields of expertise and routinely collaborate, teach and lecture at leading medical centers throughout the world. Israel also has free trade agreements with the United States and the European Union.
| 64 |
Other Countries
In addition to regulations in the United States, the EU and Israel, we are subject to a variety of other regulations governing clinical trials and commercial sales and distribution of drugs in other countries. Whether or not our products receive approval from the FDA, approval of such products must be obtained by the comparable regulatory authorities of countries other than the United States before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials and product licensing vary greatly from country to country.
The requirements that we and our collaborators must satisfy to obtain regulatory approval by government agencies in other countries prior to commercialization of our products in such countries can be rigorous, costly and uncertain. In Canada and Australia, regulatory requirements and approval processes are similar in principle to those in the United States. For example, in Canada, pharmaceutical product candidates are regulated by the Food and Drugs Act and the rules and regulations promulgated thereunder, which are enforced by the Therapeutic Products Directorate of Health Canada, or Health Canada. Before commencing clinical trials in Canada, an applicant must complete preclinical studies and file a clinical trial application with Health Canada. After filing a clinical trial application, the applicant must receive different clearance authorizations to proceed with Phase 1 clinical trials, which can then lead to Phase 2 and Phase 3 clinical trials. To obtain regulatory approval to commercialize a new drug in Canada, a new drug submission, or NDS, must be filed with Health Canada. If the NDS demonstrates that the product was developed in accordance with the regulatory authorities’ rules, regulations and guidelines and demonstrates favorable safety and efficacy and receives a favorable risk/benefit analysis, Health Canada issues a notice of compliance which allows the applicant to market the product. Facilities, procedures, operations and/or testing of products are subject to periodic inspection by Health Canada and the Health Products and Food Branch Inspectorate. In addition, Health Canada conducts pre-approval and post-approval reviews and plant inspections to determine whether systems are in compliance with the good manufacturing practices in Canada, Drug Establishment Licensing requirements and other provisions of the Food and Drug Regulations.
Foreign governments also have stringent post-approval requirements including those relating to manufacture, labeling, reporting, record keeping and marketing. Failure to substantially comply with these on-going requirements could lead to government action against the product, our company and/or our representatives.
Related Matters
From time to time, legislation is drafted, introduced and passed in governmental bodies that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA, EMA, the Israeli Ministry of Health and other applicable regulatory bodies to which we are subject. In addition, regulations and guidance are often revised or reinterpreted by the national agency in ways that may significantly affect our business and our product candidates. It is impossible to predict whether such legislative changes will be enacted, whether FDA, EMA or Israeli Ministry of Health regulations, guidance or interpretations will change, or what the impact of such changes, if any, may be. We may need to adapt our business and product candidates and products to changes that occur in the future.
C. Organizational Structure
Our corporate structure consists of Can-Fite and three subsidiaries, one of which is an indirect subsidiary: Ultratrend Limited, an English limited company, OphthaliX Inc., a Delaware corporation, or OphthaliX, and Eye-Fite Limited, an Israeli limited company, or Eye-Fite. Ultratrend Limited is a wholly-owned subsidiary of Can-Fite, but has yet to conduct any significant activity. Can-Fite holds 82% of the issued and outstanding capital stock of OphthaliX and accordingly may appoint all members of the Board of Directors of OphthaliX. Eye-Fite, a wholly-owned subsidiary of OphthaliX, holds an exclusive license from Can-Fite.
D. Property, Plants and Equipment.
We are headquartered in Petah-Tikva, Israel. We lease one floor in one facility pursuant to a lease agreement with Eshkolit Nihul Nadlan LTD, an Israeli limited company, that pursuant to a verbal agreement expires on December 31, 2017. The Petah-Tikva headquarters consists of approximately 300 square meters of space with eight parking spaces. Lease payments are approximately NIS 20,447, or $5,318, per month. If our lease is terminated, we do not foresee significant difficulty in leasing another suitable facility. The current facility houses both our administrative, clinical and research operations. The research laboratory consists of approximately 150 square meters and includes a tissue culture laboratory and a molecular biology laboratory.
| 65 |
ITEM 4A. Unresolved Staff Comments
Not Applicable.
ITEM 5. Operating and Financial Review and Prospects
The information in this section should be read in conjunction with our consolidated financial statements and related notes beginning on page F-1 and the related information included elsewhere in this Annual Report on Form 20-F. Our financial statements are prepared in accordance with IFRS as issued by the International Accounting Standards Board, and reported in NIS. We maintain our accounting books and records in NIS and our functional currency is NIS. Certain amounts presented herein may not sum due to rounding.
Overview
We are a clinical-stage biopharmaceutical company focused on developing orally bioavailable small molecule therapeutic products for the treatment of autoimmune-inflammatory indications, oncology and liver diseases as well as sexual dysfunction. Our platform technology utilizes the Gi protein associated A3AR as a therapeutic target. A3AR is highly expressed in inflammatory and cancer cells, and not significantly expressed in normal cells, suggesting that the receptor could be a unique target for pharmacological intervention. Our pipeline of drug candidates are synthetic, highly specific agonists and allosteric modulators, or ligands or molecules that initiate molecular events when binding with target proteins, targeting the A3AR.
Our strategy is to build a fully integrated biotechnology company that discovers, in-licenses and develops an innovative and effective small molecule drug portfolio of ligands that bind to a specific therapeutic target for the treatment of autoimmune-inflammatory, oncological, ophthalmic diseases and more. We continue to develop and test our existing pipeline, while also testing other indications for our existing drug candidates and examining, from time to time, the potential of other small molecules that may fit our platform technology of utilizing small molecules to target the A3AR. We generally focus on drugs with global market potential and we seek to create global partnerships to effectively assist us in developing our portfolio and to market our products.
We have in-licensed an allosteric modulator of the A3AR, CF602 from Leiden University. In addition, we have out-licensed Piclidenoson (i) for the treatment of RA to Kwang Dong Pharmaceutical Co. Ltd., a South Korean limited company, or KD for the Korean market, (ii) for the treatment of psoriasis and RA to Cipher Pharmaceuticals for the Canadian market, and (iii) for the treatment of ophthalmic diseases to Eyefite, a wholly-owned subsidiary of OphthaliX, for the global market. See “Item 4. Information on the Company—Business Overview—Out-Licensing and Distribution Agreements”.
With respect to Namodenoson, in October 2016, we entered into an exclusive distribution agreement with Chong Kun Dang Pharmaceuticals, or CKD for the exclusive right to distribute Namodenoson for the treatment of liver cancer in South Korea, upon receipt of regulatory approvals. The distribution agreement provides for up to $3,000,000 in upfront and milestone payments, plus royalties on net sales of 23%. The distribution agreement further provides that we will deliver finished product to CKD and grant CKD a right of first refusal to distribute Namodenoson for other indications for which we develop Namodenoson. See “Item 4. Information on the Company—Out-Licensing and Distribution Agreements—CKD Agreement”.
In July 2016, OphthaliX released top-line results from its Phase II clinical trial of Piclidenoson for the treatment of glaucoma. In this trial, no statistically significant differences were found between the Piclidenoson treated group and the placebo group in the primary endpoint of lowering intra ocular pressure, or IOP. High IOP is a characteristic of glaucoma. Piclidenoson was found to have a favorable safety profile and was well tolerated. Based on these overall results, OphthaliX sees no immediate path forward in glaucoma. As of the date hereof, OphthaliX has no active business operations. See “Item 10. Additional Information—Material Contracts—OphthaliX Agreements.”
In June 2015, we received a lawsuit, filed with the District Court of Tel-Aviv, requesting recognition of this lawsuit as a class action. The lawsuit named us, our Chief Executive Officer and directors as defendants. The lawsuit alleges, among other things, that we misled the public with regard to disclosures concerning the efficacy of our drug candidate, CF101 in relation to the Psoriasis studies. The claimant alleges that he suffered personal damages of over NIS 73,000, while also claiming that our shareholders suffered aggregate damages of approximately NIS 125 million. On March 31, 2016, we filed a response to the lawsuit. On March 1, 2017, a hearing was held in the District Court on whether to certify the lawsuit as a class action. A final hearing on the certification is scheduled for April 26, 2017.
| 66 |
Our product candidates, Piclidenoson, Namodenoson and CF602 are being developed to treat several autoimmune-inflammatory, oncological and sexual dysfunction indications. Piclidenoson is in various stages of clinical development for the treatment of autoimmune-inflammatory diseases, including RA and psoriasis. Namodenoson is being developed for the treatment of HCC and has orphan drug designation for the treatment of HCC in the U.S. and Europe. Recently, Namodenoson was granted Fast Track designation by the FDA as a second line treatment to improve survival for patients with advanced hepatocellular carcinoma who have previously received Nexavar (sorafenib). Namodenoson is also being developed for the treatment of non-alcoholic steatohepatitis, or NASH, following our recently concluded study which revealed compelling pre-clinical data on Namodenoson in the treatment of NASH, a disease for which no FDA approved therapies currently exist. CF602 is our second generation allosteric drug candidate for the treatment of sexual dysfunction, which has shown proof of concept in pre-clinical pharmacological studies. Preclinical studies revealed that our drug candidates have potential to treat additional inflammatory diseases, such as Crohn’s disease, oncological diseases and viral diseases, such as the JC virus.
We are currently: (i) conducting preparatory work for a Phase III trial for Piclidenoson in the treatment of RA, following agreement with the EMA on our protocol design and expect to commence enrollment in the second quarter of 2017, (ii) conducting preparatory work for a Phase III trial for Piclidenoson in the treatment of psoriasis following agreement with the EMA on our protocol design and expect IRB submissions in the fourth quarter of 2017, (iii) conducting a Phase II study with respect to the development of CF102 for the treatment of HCC and anticipate completing enrollment of approximately 78 patients during the first half of 2017, (iv) conducting preparatory work for a Phase II trial of CF102 in the treatment of NASH, a new indication identified by us for our liver cancer drug, following approval of the study protocol by IRBs and anticipate commencing enrollment in the second quarter of 2017, and (v) conducting efficacy and safety IND enabling studies with two additional compounds that belong to the family of allosteric molecules, similar to CF602, for the treatment of sexual dysfunction.
Since inception, we have incurred significant losses in connection with our research and development. At December 31, 2016, we had an accumulated deficit of approximately NIS 350 million ($91 million). Although we have recognized revenues in connection with our existing out-licensing agreements with KD, Cipher and CKD and our historic out-licensing agreement with SKK, we expect to generate losses in connection with the research and development activities relating to our pipeline of drug candidates. Such research and development activities are budgeted to expand over time and will require further resources if we are to be successful. As a result, we expect to incur operating losses, which may be substantial over the next several years, and we will need to obtain additional funds to further develop or research and development programs.
We have funded our operations primarily through the sale of equity securities (both in private placements and in public offerings) and payments received under our existing out-licensing agreements with KD, Cipher and CKD and our historic out-licensing agreement with SKK. We expect to continue to fund our operations over the next several years through our existing cash resources, potential future milestone payments that we expect to receive from our licensees, interest earned on our investments, if any, and additional capital to be raised through public or private equity offerings or debt financings. As of December 31, 2016, we had approximately $8 million, or NIS 31 million, of cash and cash equivalents based on the exchange rate reported by the Bank of Israel as of December 31, 2016.
Revenues
Our revenues to date have been generated primarily from payments under our existing out-licensing agreements with KD, Cipher and CKD and our historic out-licensing agreement with SKK.
Under the Kwang Dong Agreement, we are entitled to up-front and milestone payments of up to $1.5 million. In accordance with the Kwang Dong Agreement, we received an up-front payment of $0.3 million and a payment of $0.048 million as consideration for KD’s purchase of our ordinary shares in 2009 and a milestone payment of $0.2 million in 2010. Under the terms of the Kwang Dong Agreement, in addition to the payments mentioned above, we are entitled to certain additional payments based on the sale of raw materials, subject to the terms and conditions of the respective agreements. See “Item 4. Information on the Company—Business Overview—Out-Licensing and Distribution Agreements”.
| 67 |
Under the Distribution and Supply Agreement with Cipher we received CDN$1.65 million upon execution of the agreement and are entitled to milestone payments upon receipt of regulatory approval by Health Canada for Piclidenoson and the first delivery of commercial launch quantities as follows (i) CDN$1 million upon the first approved indication for either psoriasis or RA, and (ii) CDN $1 million upon the second approved indication for either psoriasis or RA. In addition, following regulatory approval, we shall be entitled to a royalty of 16.5% of net sales of Piclidenoson in Canada and reimbursement for the cost of manufacturing Piclidenoson. We are also entitled to a royalty payment for any authorized generic of Piclidenoson that Cipher distributes in Canada. See “Item 4. Information on the Company—Business Overview—Out-Licensing and Distribution Agreements”.
The Distribution Agreement with CKD provides for up to $3,000,000 in upfront and milestone payments payable as follows: (i) an upfront payment of $500,000 within 30 days of receipt of an invoice from us, which we received in the fourth quarter of 2016 and (ii) within 30 days of the occurrence of each of the following: (1) $500,000 upon receipt by CKD of a positive result from the preliminary review by the Ministry of Food and Drug Safety, or MFDS, on obtaining orphan drug designation for Namodenoson in South Korea, (2) $500,000 upon successful completion of our ongoing Phase II clinical trial for Namodenoson, (3) $1,000,000 upon the granting of marketing authorization of Namodenoson in South Korea by the MFDS, and (4) $500,000 upon registration of Namodenoson on the “reimbursement listing” in South Korea by the National Health Insurance Services in Korea. In addition, we are entitled to a royalty of 23% of net sales of Namodenoson following commercial launch in South Korea which includes the transfer price for delivering finished product to CKD. See “Item 4. Information on the Company—Business Overview—Out-Licensing and Distribution Agreements”.
Under the terminated SKK license agreement we received an aggregate of approximately $8.5 million from SKK. See “Item 4. Information on the Company—Business Overview—Out-Licensing and Distribution Agreements”.
Certain payments we have received from SKK and KD have been subject to a 10% and 5% withholding tax in Japan and Korea, respectively, and certain payments we may receive in the future, if at all, may also be subject to the same withholding tax in Korea. Receipt of any milestone payment under our out-licensing agreements depends on many factors, some of which are beyond our control. We cannot assure you that we will receive any of these future payments. We expect our revenues for the next several years, if any, to be derived primarily from payments under our current out-license agreements and our public capital raising activities, as well as additional collaborations that we may enter into in the future with respect to our drug candidates.
Research and Development
Our research and development expenses consist primarily of salaries and related personnel expenses, fees paid to external service providers, up-front and milestone payments under our license agreements, patent-related legal fees, costs of preclinical studies and clinical trials, drug and laboratory supplies and costs for facilities and equipment. We charge all research and development expenses to operations as they are incurred. We expect our research and development expense to remain our primary expense in the near future as we continue to develop our products. Increases or decreases in research and development expenditures are attributable to the number and/or duration of the pre-clinical and clinical studies that we conduct.
The following table identifies our current major research and development projects:
| Project | Status | Expected or Recent Near Term Milestone | ||||
| CF 101 | Preparing for a Phase III study in RA | Expect to commence enrollment in 2017 | ||||
| Preparing for a Phase III study in psoriasis | Expect to commence enrollment in 2017 | |||||
| CF 102 | Phase II in HCC | Completion of patient enrollment in first half of 2017 | ||||
| Preparing for a Phase II study in NASH | Expect to commence enrollment in second quarter of 2017 | |||||
| CF 602 | Conducting IND enabling studies | Planning to file an IND with the FDA in 2017 |
We record certain costs for each development project on a “direct cost” basis, as they are recorded to the project for which such costs are incurred. Such costs include, but are not limited to, CRO expenses, drug production for pre-clinical and clinical studies and other pre-clinical and clinical expenses. However, certain other costs, including but not limited to, salary expenses (including salaries for research and development personnel), facilities, depreciation, share-based compensation and other overhead costs are recorded on an “indirect cost” basis, i.e., they are shared among all of our projects and are not recorded to the project for which such costs are incurred. We do not allocate direct salaries to projects due to the fact that our project managers are generally involved in several projects at different stages of development, and the related salary expense is not significant to the overall cost of the applicable projects. In addition, indirect labor costs relating to our support of the research and development process, such as manufacturing, controls, pre-clinical analysis, laboratory testing and initial drug sample production, as well as rent and other administrative overhead costs, are shared by many different projects and have never been considered by management to be of significance in its decision-making process with respect to any specific project. Accordingly, such costs have not been specifically allocated to individual projects.
| 68 |
Set forth below is a summary of the gross direct costs allocated to our main projects on an individual basis, as well as the gross direct costs allocated to our less significant projects on an aggregate basis, for the years ended December 31, 2014, 2015 and 2016; and on an aggregate basis since project inception:
| (USD in thousands) | Total Costs | |||||||||||||||
| Year Ended December 31, | Since Project | |||||||||||||||
| 2014 | 2015 | 2016 | Inception | |||||||||||||
| Piclidenoson | 1,866 | 971 | 1,946 | 21,481 | ||||||||||||
| Namodenoson | 1,289 | 1,044 | 1,907 | 5,628 | ||||||||||||
| CF602 | 23 | 243 | 1,126 | 1,392 | ||||||||||||
| Other projects | 18 | 1 | - | 1,729 | ||||||||||||
| Total gross direct project costs (1) | 3,196 | 2,259 | 4,979 | 30,230 | ||||||||||||
| (1) | Does not include indirect project costs and overhead, such as payroll and related expenses (including stock-based compensation), facilities, depreciation and impairment of intellectual property, which are included in total research and development expenses in our financial statements. |
From our inception through December 31, 2016, we have incurred research and development expenses of approximately $84 million. We expect that a large percentage of our research and development expense in the future will be incurred in support of our current and future preclinical and clinical development projects. Due to the inherently unpredictable nature of preclinical and clinical development processes and given the early stage of our preclinical product development projects, we are unable to estimate with any certainty the costs we will incur in the continued development of the product candidates in our pipeline for potential commercialization. Clinical development timelines, the probability of success and development costs can differ materially from expectations. We expect to continue to test our product candidates in preclinical studies for toxicology, safety and efficacy, and to conduct additional clinical trials for each product candidate. If we are not able to enter into an out-licensing arrangement with respect to any product candidate prior to the commencement of later stage clinical trials, we may fund the trials for the product candidates ourselves.
While we are currently focused on advancing each of our product development projects, our future research and development expenses will depend on the clinical success of each product candidate, as well as ongoing assessments of each product candidate’s commercial potential. In addition, we cannot forecast with any degree of certainty which product candidates may be subject to future out-licensing arrangements, when such out-licensing arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements.
As we obtain results from clinical trials, we may elect to discontinue or delay clinical trials for certain product candidates or projects in order to focus our resources on more promising product candidates or projects. Completion of clinical trials by us or our licensees may take several years or more, but the length of time generally varies according to the type, complexity, novelty and intended use of a product candidate.
The cost of clinical trials may vary significantly over the life of a project as a result of differences arising during clinical development, including, among others:
| ● | the number of sites included in the clinical trials; |
| ● | the length of time required to enroll suitable patients; |
| ● | the number of patients that participate in the clinical trials; |
| ● | the duration of patient follow-up; |
| ● | the development stage of the product candidate; and |
| ● | the efficacy and safety profile of the product candidate. |
| 69 |
We expect our research and development expenses to increase in the future from current levels as we continue the advancement of our clinical trials and preclinical product development and to the extent we in-license new product candidates. The lengthy process of completing clinical trials and seeking regulatory approval for our product candidates requires expenditure of substantial resources. Any failure or delay in completing clinical trials, or in obtaining regulatory approvals, could cause a delay in generating product revenue and cause our research and development expenses to increase and, in turn, have a material adverse effect on our operations. Because of the factors set forth above, we are not able to estimate with any certainty when we would recognize any net cash inflows from our projects.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation for employees in executive and operational functions, including accounting, finance, legal, business development, investor relations, information technology and human resources. Other significant general and administration costs include facilities costs, professional fees for outside accounting and legal services, travel costs, insurance premiums and depreciation.
Financial Expense and Income
Financial expense and income consists of interest earned on our cash and cash equivalents; bank fees and other transactional costs; expense or income resulting from fluctuations of the U.S. dollar and other currencies, in which a portion of our assets and liabilities are denominated, against the NIS (our functional currency); and fluctuations in the market value of our U.S. dollar warrants.
Critical Accounting Policies and Estimates
Our accounting policies and their effect on our financial condition and results of operations are more fully described in our audited consolidated financial statements included elsewhere in this Annual Report. The preparation of financial statements in conformity with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB, requires management to make estimates and assumptions that in certain circumstances affect the reported amounts of assets and liabilities, revenues and expenses and disclosure of contingent assets and liabilities. These estimates are prepared using our best judgment, after considering past and current events and economic conditions. While management believes the factors evaluated provide a meaningful basis for establishing and applying sound accounting policies, management cannot guarantee that the estimates will always be consistent with actual results. In addition, certain information relied upon by us in preparing such estimates includes internally generated financial and operating information, external market information, when available, and when necessary, information obtained from consultations with third party experts. Actual results could differ from these estimates and could have a material adverse effect on our reported results.
We believe that the accounting policies discussed below are critical to our financial results and to the understanding of our past and future performance, as these policies relate to the more significant areas involving management’s estimates and assumptions. We consider an accounting estimate to be critical if: (1) it requires us to make assumptions because information was not available at the time or it included matters that were highly uncertain at the time we were making our estimate; and (2) changes in the estimate could have a material impact on our financial condition or results of operations.
| 70 |
Functional Currency
The presentation currency of our financial statements and our functional currency is the NIS. The functional currency of an entity in which we own an equity interest, which is referred to as oursubsidiary, differs from our functional currency, that subsidiary represents a foreign operation whose financial statements are translated as follows: (i) assets and liabilities are translated at the closing rate at the date of that balance sheet, (ii) income and expenses are translated at average exchange rates for the presented periods and (iii) share capital and capital reserves are translated at the exchange rate prevailing at the date of incurrence. All resulting translation differences are recognized in a separate component in equity, as other comprehensive loss, “adjustments from translation of financial statements.”
For the convenience of the reader, the reported NIS amounts as of December 31, 2016 have been translated into U.S. dollars at the representative rate of exchange on December 31, 2016 (U.S. $1 = NIS 3.845). The U.S. dollar amounts presented should not be construed as representing amounts that are receivable or payable in U.S. dollars or convertible into U.S. dollars, unless otherwise indicated. The U.S. dollar amounts were rounded to whole numbers of convenience.
Principles of Consolidation
Our financial statements reflect the consolidation of the financial statements of companies that we control based on legal control or effective control. We fully consolidate into our financial statements the results of operations of companies that we control. Legal control exists when we have the power, directly or indirectly, to govern the financial and operating policies of an entity. The effect of potential voting rights that are exercisable at the balance sheet date are considered when assessing whether we have legal control. In addition, we consolidate on the basis of effective control even if we do not have voting control. The determination that effective control exists involves significant judgment.
In evaluating the effective control on our investees we consider the following criteria to determine if effective control exists:
| ● | whether we hold a significant voting interest (but less than half the voting rights); | |
| ● | whether there is a wide diversity of public holdings of the remaining shares conferring voting rights; | |
| ● | whether in the past we had the majority of the voting power participating in the general meetings of shareholders and, therefore, have in fact had the right to nominate the majority of the board members; | |
| ● | the absence of a single entity that holds a significant portion of the investee’s shares; | |
| ● | our ability to establish policies and guide operations by appointing the remainder of the investee’s senior management; and | |
| ● | whether the minority shareholders have participation rights or other preferential rights, excluding traditional shareholder protective rights. | |
Entities we control are fully consolidated in our financial statements. All significant intercompany balances and transactions are eliminated in consolidation. Non-controlling interests of subsidiaries represent the non-controlling shareholders’ proportionate interest in the comprehensive income (loss) of the subsidiaries and fair value of the net assets or the net identifiable assets upon the acquisition of the subsidiaries.
Revenue Recognition
We generate income from distribution agreements. See “Item 4. Information on the Company—Business Overview—Out-Licensing and Distribution Agreements”. Such income comprises of upfront license fees, milestone payments and potential royalty payments.
We identified four components in the agreements: (i) performing the research and development services through regulatory approval; (ii) exclusive license to distribute; (iii) participation in joint steering committee; and, (iv) royalties resulting from future sales of the product.
We recognize revenue in accordance with IAS 18, "Revenue" pursuant to which each required deliverable is evaluated to determine whether it qualifies as a separate unit of accounting based on whether the deliverable has "stand-alone value" to the customer. The arrangement’s consideration that is fixed or determinable is then allocated to each separate unit of accounting based on the relative selling price of each deliverable which is based on the Estimated Selling Price.
| 71 |
Components (i) – (iii) were analyzed as one unit of accounting. Consequently, revenue from these components is recorded based on the term of the research and development services (which is the last deliverable in the arrangement). We estimate these services will spread over a period of 24 quarters beginning March 2015.
Revenues from milestone payments:
Contingent payments related to milestones will be recognized immediately upon satisfaction of the milestone and contingent payments related to royalties will be recognized in the period that the related sales have occurred.
Revenues from royalties:
Revenues from royalties will be recognized as they accrue in accordance with the terms of the relevant agreement.
Share-based Compensation
We account for share-based compensation arrangements in accordance with the provisions of IFRS 2. IFRS 2 requires companies to recognize share-based compensation expense for awards of equity instruments based on the grant-date fair value of those awards. The cost is recognized as compensation expense over the vesting period, based upon the grant-date fair value of the equity or liability instruments issued. We selected the binomial option pricing model as the most appropriate method for determining the estimated fair value of our share-based awards without market conditions. The determination of the grant date fair value of options using an option pricing model is affected by estimates and assumptions regarding a number of complex and subjective variables. These variables include the expected volatility of our share price over the expected term of the options, share option exercise and forfeiture rate, risk-free interest rates, expected dividends and the price of our ordinary shares on the TASE. As our ordinary shares are publicly traded on the TASE, we do not need to estimate the fair value of our ordinary shares. Rather, we use the actual closing market price of our ordinary shares on the date of grant, as reported by the TASE although in the future may use the closing market price of our ADSs on the date of grant, as reported by the NYSE MKT.
If any of the assumptions used in the binomial option pricing model change significantly, share-based compensation for future awards may differ materially compared with the awards previously granted.
As for other service providers, the cost of the transactions is measured at the fair value of the goods or services received as consideration for equity instruments. In cases where the fair value of the goods or services received as consideration of equity instruments cannot be measured, they are measured by reference to the fair value of the equity instruments granted.
The cost of equity-settled transactions is recognized in profit or loss, together with a corresponding increase in equity, during the period which the service are to be satisfied, ending on the date on which the relevant employees or other service providers become fully entitled to the award.
If we modify the conditions on which equity-instruments are granted, an additional expense is recognized for any modification that increases the total fair value of the share-based payment arrangement or is otherwise beneficial to the employee or other service provider at the modification date.
Liability Related to Certain Warrants
The fair value of the liability for warrants exercisable into shares issued to investors in connection with our financings to date was calculated using the Black-Scholes-Merton option-pricing model. We accounted for these warrants as liabilities due to the dollar exercise price terms and in accordance with IAS 39, measured at fair value each reporting period until they will be exercised or expired, with changes in the fair values being recognized in our statement of comprehensive loss as financial income or expense.
Fair value for each reporting period was calculated based on the following assumptions:
| 1. | Risk-free interest rate - based on yield rated of non-index linked U.S. Federal Reserve treasury bonds. | ||
| 2. | Expected volatility - was calculated based on our actual historical stock price movements over a term that is equivalent to the expected term of the option. | ||
| 3. | Expected life - the expected life was based on the expiration date of the warrants. | ||
| 4. | Expected dividend yield - was based on the fact that we have not paid dividends to its shareholders in the past and does not expect to pay dividends to its shareholders in the future. |
Our net loss for the year ended December 31, 2016 and 2015 included finance income in the amount of NIS 27,009,000 and NIS 19,770,000, respectively, in connection with the above-mentioned warrants.
| 72 |
Recently Issued Accounting Pronouncements
IFRS 9-Financial Instruments:
In July 2014, the IASB issued the final version of IFRS 9 Financial Instruments which reflects all phases of the financial instruments project and replaces IAS 39 Financial Instruments: Recognition and Measurement and all previous versions of IFRS 9. The standard introduces new requirements for classification and measurement, impairment, and hedge accounting. IFRS 9 is effective for annual periods beginning on or after 1 January 2018, with early application permitted. The adoption of IFRS 9 will have no material effect on the Company’s financial assets on the financial statements.
IAS 7-Statement of Cash Flows:
In January 2016, the IASB issued amendments to IAS 7 Statement of Cash Flows. This standard requires additional disclosures regarding financial liabilities. The amendments are effective for annual periods beginning on or after January 1, 2017, with early application permitted. We plan to include the necessary disclosures in the financial statements when applicable.
IFRS 15-Revenue from Contracts with Customers:
In May 2014, the IASB issued IFRS 15 Revenue from Contracts with Customers, which replaces IAS 18 Revenue, IAS 11 Construction Contracts, IFRIC 13 Customer Loyalty Programs, IFRIC 15 Agreements for the Construction of Real Estate, IFRIC 18 Transfers of Assets from Customers and SIC-31 Revenue - Barter Transactions Involving Advertising Services. IFRS 15 is to be applied retrospectively for annual periods beginning on or after January 1, 2018 with early adoption permitted. We are evaluating the possible impact of IFRS 15 but are presently unable to assess its effect, if any, on the financial statements.
IFRS 16-Leases:
IFRS 16 replaces International Accounting Standard 17 - Leases (IAS 17) and its related interpretations. The standard's instructions annul the existing requirement from lessees to classify leases as operating or finance leases. Instead of this, for lessees, the new standard presents a unified model for the accounting treatment of all leases according to which the lessee has to recognize an asset and liability in respect of the lease in its financial statements. Similarly, the standard determines new and expanded disclosure requirements from those required at present. The standard will become effective for annual periods as of January 1, 2019, with early adoption permitted. We have not yet commenced examining the effects of adopting the standard on the financial statements.
U.S. Offerings
On March 10, 2014, we sold to accredited investors 982,344 ADSs, at a purchase price of $5.15 per ADS, and warrants to purchase 491,172 additional ADSs in a private placement resulting in gross proceeds of $5,059,072. The warrants may be exercised at any time after September 10, 2014 for a period of four years from the date of issuance and have an exercise price of $6.43 per ADS, subject to adjustment as set forth therein. The warrants may be exercised on a cashless basis if after September 10, 2014 there is no effective registration statement registering the ADSs underlying the warrants. In connection with the private placement we paid an aggregate of $509,840 in placement agent fees and expenses and we issued placement agent warrants to purchase 49,117 ADSs exercisable at $6.43 per ADS for four years. The placement agent warrants may be exercised on a cashless basis at any time after September 10, 2014.
On December 8, 2014, we sold to certain institutional investors an aggregate of 1,797,753 ADSs in a registered direct offering at $4.45 per share resulting in gross proceeds of $8,000,000. In addition, we issued to the investors unregistered warrants to purchase 898,877 ADSs in a private placement. The warrants may be exercised at any time for a period of five years from issuance and have an exercise price of $4.45 per ADS, subject to adjustment as set forth therein. The warrants may be exercised on a cashless basis if six months after issuance there is no effective registration statement registering the ADSs underlying the warrants. We paid an aggregate of $762,500 in placement agent fees and expenses and issued unregistered placement agent warrants to purchase 89,888 ADS, exercisable for five years from issuance, at an exercise price of $4.45 per ADS, subject to adjustment as set forth therein.
| 73 |
On September 21, 2015, we sold to certain institutional investors an aggregate of 2,068,966 ADSs in a registered direct offering at $4.35 per ADS resulting in gross proceeds of $9,000,002. In addition, we issued to the investors unregistered warrants to purchase 1,034,483 ADSs in a private placement. The warrants may be exercised after six months from issuance for a period of five and a half years from issuance and have an exercise price of $5.25 per ADS, subject to adjustment as set forth therein. The warrants may be exercised on a cashless basis if six months after issuance there is no effective registration statement registering the ADSs underlying the warrants. We paid an aggregate of $792,379 in placement agent fees and expenses and issued unregistered placement agent warrants to purchase 103,448 ADS on the same terms as the warrants except they have a term of five years.
On October 15, 2015, we sold to certain institutional investors providing for the issuance of an aggregate of 1,109,196 ADSs in a registered direct offering at $4.35 per ADS resulting in gross proceeds of approximately $4,825,000. In addition, we issued to the investors unregistered warrants to purchase 443,678 ADSs in a private placement. The warrants may be exercised after six months from issuance for a period of five and a half years from issuance and have an exercise price of $5.25 per ADS, subject to adjustment as set forth therein. The warrants may be exercised on a cashless basis if six months after issuance there is no effective registration statement registering the ADSs underlying the warrants. We paid an aggregate of $524,621 in placement agent fees and expenses and issued unregistered placement agent warrants to purchase 55,460 ADS on the same terms as the warrants except they have a term of five years.
On January 24, 2017, we sold to certain institutional investors providing for the issuance of an aggregate of 2,500,000 ADSs in a registered direct offering at $2.00 per ADS resulting in gross proceeds of approximately $5,000,000. In addition, we issued to the investors unregistered warrants to purchase 1,250,000 ADSs in a private placement. The warrants may be exercised after six months from issuance for a period of five and a half years from issuance and have an exercise price of $2.25 per ADS, subject to adjustment as set forth therein. The warrants may be exercised on a cashless basis if six months after issuance there is no effective registration statement registering the ADSs underlying the warrants. We paid an aggregate of $360,000 in placement agent fees and expenses and issued unregistered placement agent warrants to purchase 125,000 ADS on the same terms as the warrants except they have a term of five years.
Israeli Public Warrant Offerings
Series 6 and 7 Warrants
In connection with our Israeli public offering on November 16, 2011, we issued Series 6 and Series 7 Warrants, which were publicly traded on the TASE and exercisable into our publicly traded ordinary shares. In accordance with IFRS, we allocated a portion of the consideration received for such warrants based on their market value at that time. The consideration allocated to such warrants is generally reflected in non-current liabilities due to the fact that the exercise price of the warrants is linked to the Israeli consumer price index.
In the public offering, we issued 4,953,750 Series 6 Warrants exercisable for 198,150 of our ordinary shares. The Series 6 Warrants had an exercise price of NIS 15.75 per ordinary share (which may fluctuate as it is based on the Israeli consumer price index) and were originally scheduled to expire on May 16, 2012. On August 18, 2012, we filed an application with the Petah-Tikva District Court in Israel to approve an extension of the Series 6 Warrants until September 1, 2014 and following a meeting of our shareholders and holders of Series 6 Warrant to approve the extension of the exercise period of the Series 6 Warrants, on January 27, 2014, the District Court approved the extension until October 30, 2013. The Series 6 Warrants expired on October 30, 2013.
In the same offering, we issued 9,907,500 Series 7 Warrants exercisable for 396,300 of our ordinary shares. The Series 7 Warrants had an exercise price of NIS 20 per ordinary share (which may fluctuate as it is based on the Israeli consumer price index) and were originally scheduled to expire on November 16, 2013. On November 7, 2013, we filed an application with the Petah-Tikva District Court in Israel to approve an extension of the Series 7 Warrants until March 31, 2014 and following a meeting of our shareholders and holders of Series 7 Warrant to approve the extension of the exercise period of the Series 7 Warrants, on January 27, 2014, the District Court approved the extension until March 31, 2014. The Series 7 warrants expired on March 31, 2014.
| 74 |
Series 8 and 9 Warrants
In connection with our Israeli public offering on May 1, 2012, we issued Series 8 and Series 9 Warrants, which were publicly traded on the TASE and exercisable into our publicly traded ordinary shares. In accordance with IFRS, we allocated a portion of the consideration received for such warrants based on their market value at the time. The consideration allocated to warrants is generally reflected in non-current liabilities due to the fact that the exercise price of such warrants is linked to the Israeli consumer price index.
We issued 8,112,000 Series 8 Warrants exercisable for 324,480 of our ordinary shares in the offering. Although the Series 8 Warrants had an exercise price of NIS 13.75 per ordinary share (which may fluctuate as it is based on the Israeli consumer price index) and were set to expire on June 30, 2013, on June 24, 2013, the Lod District Court in Israel approved a settlement, approved at a meeting of the shareholders and the Series 8 Warrants holders, according to which the exercise price was increased to 18.75 NIS per ordinary share (which may fluctuate as it is based on the Israeli consumer price index) and the exercise period was extended until December 31, 2013.The Series 8 Warrants expired on December 31, 2013.
We also issued 12,168,000 Series 9 Warrants exercisable for 486,720 of our ordinary shares in this offering. In accordance with IFRS, we allocated a portion of the consideration received from the Series 9 Warrants based on their market value at the time. The consideration allocated to the Series 9 Warrants is generally reflected in shareholders’ equity due to the fact that the exercise price of such warrants is fixed. The Series 9 Warrants had a fixed exercise price of NIS 21.25 per ordinary share. The Series 9 Warrants expired on May 1, 2015.
Series 10 and 11 Warrants
In connection with our Israeli public offering on February 5, 2013, we issued Series 10 and Series 11 Warrants, which are publicly traded on the TASE and exercisable into our publicly traded ordinary shares. In accordance with IFRS, we allocated a portion of the consideration received for such warrants based on their market value at the time. The consideration allocated to warrants is generally reflected in non-current liabilities due to the fact that the exercise price of such warrants is linked to the Israeli consumer price index.
We issued 39,067,000 Series 10 Warrants exercisable for 1,562,680 of our ordinary shares in the offering. The Series 10 Warrants had an exercise price of NIS 0.394 per ordinary share (which may fluctuate as it is based on the Israeli consumer price index) and following extension were set to expire on October 31, 2016. On October 10, 2016, we filed an application with the Lod District Court in Israel to approve an extension of the Series 11 Warrants until October 31, 2017. Following a meeting of our shareholders and holders of Series 11 Warrant, on January 4, 2017 the District Court approved the extension until October 31, 2017.
We also issued 37,385,000 Series 11 Warrants exercisable for 1,495,400 of our ordinary shares in the offering. The Series 11 Warrants have an exercise price of NIS 0.392 per ordinary share (which may fluctuate as it is based on the Israeli consumer price index) and were set to expire on April 30, 2016. On June 14, 2016, we filed an application with the Lod District Court in Israel to approve an extension of the Series 11 Warrants until October 31, 2016 and allowing the Series 11 Warrants to be exercised on an any trading day. Following a meeting of our shareholders and holders of Series 11 Warrant, on July 13, 2016 the District Court approved the extension until October 31, 2016. On October 10, 2016, we filed an application with the Lod District Court in Israel to approve an extension of the Series 11 Warrants until October 31, 2017. Following a meeting of our shareholders and holders of Series 11 Warrant, on January 4, 2017 the District Court approved the extension until October 31, 2017.
Our Board of Directors decided that the exercise price of the Series 10 and Series 11 Warrants will no longer be linked to the Israeli consumer price index and on August 20, 2013, the Lod District Court approved a settlement, approved at a meeting of the shareholders and the Series 10 and 11Warrants holders, according to which the exercise price of the Series 10 and 11 Warrants will no longer be linked to the Israeli consumer price index. As a result, Series 10 and 11 Warrants, were reclassified to equity.
Series 12 Warrants
We issued 1,470,000 Series 12 Warrants exercisable for 1,470,000 of our ordinary shares. The Series 12 Warrants have an exercise price of NIS 15.29 per ordinary share (which may fluctuate as it is based on the Israeli consumer price index) and were originally scheduled to expire on October 22, 2016. On October 10, 2016, we filed an application with the Lod District Court in Israel to approve an extension of the Series 12 Warrants until October 31, 2017. Following a meeting of our shareholders and holders of Series 12 Warrant, on January 4, 2017 the District Court approved the extension until October 31, 2017.
As of March 29, 2017, other than Series 6, Series 7, Series 8 and Series 9 Warrants that have expired, 25,000 Series 10 Warrants to purchase 1,000 ordinary shares were exercised on December 26, 2013 for an aggregate exercise price of NIS 9,850 and 12,500 Series 11 Warrants to purchase 500 ordinary shares were exercised on December 26, 2013 for an aggregate exercise price of NIS 4,900, none of the foregoing warrants have been exercised.
Jumpstart Our Business Startups Act of 2012
We are an emerging growth company within the meaning of the rules under the Securities Act, and we will utilize certain exemptions from various reporting requirements that are applicable to public companies that are not emerging growth companies. The JOBS Act permits us, as an “emerging growth company,” to take advantage of an extended transition period to comply with certain new or revised accounting standards if such standards apply to companies that are not issuers. We are choosing to “opt out” of this provision and, as a result, we will comply with new or revised accounting standards when they are required to be adopted by issuers. This decision to opt out of the extended transition period under the JOBS Act is irrevocable.
| 75 |
A. Results of Operations
Comparison of the Year Ended December 31, 2016 to Year Ended December 31, 2015
Revenues
Revenues for the year ended December 31, 2016 were NIS 0.65 million, an increase of NIS 0.01 million, or 1.6%, compared to NIS 0.64 million for the year ended December 31, 2015. The revenues during 2016 were mainly due to the recognition of a portion of the NIS 5.14 million (CAD 1.65 million) advance payment received in March 2015 under the distribution agreement with Cipher and a minor amount due to the recognition of a portion of the NIS 1.9 million ($0.5 million) advance payment received in December 2016 under the distribution agreement with CKD.
Research and development expenses
Research and development expenses for the year ended December 31, 2016 were NIS 23.38 million, an increase of NIS 8.33 million, or 55%, compared to NIS 15.05 million for the year ended December 31, 2015. Research and developments expenses for the year ended 2016 comprised primarily of expenses associated with the Phase II study for Namodenoson as well as expenses for ongoing studies of Piclidenoson. The increase is primarily due to costs associated with preparations of the Piclidenoson Phase III studies in the treatment of RA and psoriasis and costs associated with the ongoing clinical trial of Namodenoson for treatment in liver cancer. We expect that the research and development expenses will increase through 2017 and beyond.
General and administrative expenses
General and administrative expenses were NIS 10.48 million for the year ended December 31, 2016 a decrease of NIS 0.15 million, or 1.4%, compared to NIS 10.63 million for the year ended December 31, 2015. The minor decrease is primarily due to a decrease in professional services. We expect that general and administrative expenses will remain at the same level through 2017.
Financial expenses, net
Financial income, net for the year ended December 31, 2016 aggregated NIS 6.31 million compared to financial income, net of NIS 5.29 million for the same period in 2015. The increase in financial income, net in the year ended December 31, 2016 was mainly due to fact that during 2016 we did not record any issuance expenses unlike in 2015.
Comparison of the Year Ended December 31, 2015 to Year Ended December 31, 2014
Revenues
In the year ended December 31, 2015, we recorded revenues of NIS 0.64 million. We did not record any revenues during the year ended December 31, 2014. The increase in revenue was due to the recognition of a portion of the NIS 5.14 million (CAD 1.65 million) advance payment received in March 2015 under the distribution agreement with Cipher.
Research and development expenses
Research and development expenses for the year ended December 31, 2015 were NIS 15.05 million, a decrease of NIS 1.15 million, or 7.1%, compared to NIS 16.20 million for the year ended December 31, 2014. Research and developments expenses for the year ended 2015 comprised primarily of expenses associated with the Phase II study for Namodenoson as well as expenses for ongoing studies of Piclidenoson. The decrease is primarily due to the completion of the Phase II/III psoriasis study during the first quarter of 2015 and a decrease in the scope of the non-clinical expenses during the year ended 2015 as compared to the parallel period in 2014. We expect that the research and development expenses will increase through 2016 and beyond.
| 76 |
General and administrative expenses
General and administrative expenses were NIS 10.63 million for the year ended December 31, 2015 and NIS 11.57 million for year ended December 31, 2014. The decrease is primarily due to a reduction in salary and investors and public relations expenses. We expect that general and administrative expenses will remain at the same level through 2016 and beyond.
Financial expenses, net
Financial income, net for the year ended December 31, 2015 aggregated NIS 5.29 million compared to financial income, net of NIS 3.27 million for the same period in 2014. The increase in financial income, net in the year ended 2015 was mainly due to a decrease in the fair value of warrants that are accounted as financial liability.
B. Liquidity and Capital Resources
Since inception, we have funded our operations primarily through public (in Israel and US) and private offerings of our equity securities and payments received under our strategic licensing arrangements. At December 31, 2016, we had approximately NIS 31.2 million ($8.1 million) in cash and cash equivalents, and have invested most of our available cash funds in short-term bank deposits. During the fourth quarter of 2016, we received approximately NIS 1.9 million ($0.5 million) from CKD, as upfront payment for entering into the distribution agreement with CKD. In January 2017, we raised approximately NIS 18.9 million ($5 million) in a registered direct offering.
We may be able to use U.S. taxes withheld as credits against Israeli corporate income tax when we have income, if at all, but there can be no assurance that we will be able to realize the credits. In addition, we believe that we may be entitled to a refund of such withholding tax from the U.S. government but there can be no assurance that we will be entitled to such a refund. For information regarding the revenues and expenses associated with our licensing agreements, see “Item 4. Information on the Company—Business Overview—Out-Licensing and Distribution Agreements”, “Item 4. Information on the Company—Business Overview—In-Licensing Agreements” and “Item 5. Operating and Financial Review and Prospects—Revenues.”
Net cash used in operating activities was NIS million for the year ended December 31, 2016 was NIS 34.3 million, compared with net cash used in operating activities of NIS 18.2 million and NIS 28.5 million for the years ended December 31, 2015 and 2014, respectively. The NIS 16.1 million increase in the net cash used in operating activities during 2016, compared to 2015, was primarily the result of an increase in clinical trials and increase in prepaid expenses for clinical trials supply. The NIS 10.4 million decrease in the net cash used in operating activities during 2015, compared to 2014, was primarily the result of a decrease in loss and an increase in deferred revenues.
Net cash used in investing activities for the year ended December 31, 2016 was NIS 0.04 million compared to net cash used in investing activities of NIS 0.17 million for the year ended December 31, 2015 and net cash used in investing activities of NIS 0.04 million for the year ended December 31, 2014. The changes in cash flows from investing activities are immaterial.
There was no net cash provided by financing activities for the year ended December 31, 2016, compared to net cash provided by financing activities of NIS 48.3 million for the year ended December 31, 2015 and NIS 44.7 million for the year ended December 31, 2014. The NIS 48.3 million decrease in the net cash provided by financing activities during 2016, compared to 2015, was due to no issuance of shares and warrants during 2016. The NIS 3.6 million increase in the net cash provided by financing activities during 2015, compared to 2014, was primarily due to issuance of shares and warrants, net of issuance expenses.
Developing drugs, conducting clinical trials and commercializing products is expensive and we will need to raise substantial additional funds to achieve our strategic objectives. Although we believe our existing financial resources as of March 30, 2017, will be sufficient to fund our projected cash requirements at least through the next twelve months, we will require significant additional financing to fund our operations. Additional financing may not be available on acceptable terms, if at all. Our future capital requirements will depend on many factors, including:
●
● |
the level of research and development investment required to develop our product candidates;
the failure to obtain regulatory approval or achieve commercial success of our product candidates, including Piclidenoson, Namodenoson and CF602; |
| ● | the results of our preclinical studies and clinical trials for our earlier stage product candidates, and any decisions to initiate clinical trials if supported by the preclinical results; |
| 77 |
| ● | the costs, timing and outcome of regulatory review of our product candidates that progress to clinical trials; |
| ● | the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our issued patents and defending intellectual property-related claims; |
| ● | the cost of commercialization activities if any of our product candidates are approved for sale, including marketing, sales and distribution costs; |
| ● | the cost of manufacturing our product candidates and any products we successfully commercialize; |
| ● | the timing, receipt and amount of sales of, or royalties on, our future products, if any; |
| ● | the expenses needed to attract and retain skilled personnel; |
| ● | any product liability or other lawsuits related to our products; |
| ● | the extent to which we acquire or invest in businesses, products or technologies and other strategic relationships; |
●
● |
the costs of financing unanticipated working capital requirements and responding to competitive pressures; and
maintaining minimum shareholders’ equity requirements under the NYSE MKT Company Guide. |
Until we can generate significant continuing revenues, we expect to satisfy our future cash needs through payments received under our license agreements, debt or equity financings, or by out-licensing other product candidates. We cannot be certain that additional funding will be available to us on acceptable terms, or at all. If funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts.
C. Research and Development, Patents and Licenses, Etc.
For information concerning our research and development policies and a description of the amount spent during each of the last three fiscal years on company-sponsored research and development activities, see “Item 5. Operating and Financial Review and Prospects—Operating Results.”
D. Trend Information.
We are a development stage company and it is not possible for us to predict with any degree of accuracy the outcome of our research, development or commercialization efforts. As such, it is not possible for us to predict with any degree of accuracy any significant trends, uncertainties, demands, commitments or events that are reasonably likely to have a material effect on our net sales or revenues, income from continuing operations, profitability, liquidity or capital resources, or that would cause financial information to not necessarily be indicative of future operating results or financial condition. However, to the extent possible, certain trends, uncertainties, demands, commitments and events are identified in the preceding subsections of this Item 5.
E. Off-Balance Sheet Arrangements.
We have no off-balance sheet arrangements that have had or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to investors.
| 78 |
F. Contractual Obligations.
The following table summarizes our significant contractual obligations in NIS at December 31, 2016:
| Total | Less than 1 year | 1 – 3 years | 3-5 years | More than 5 years | ||||||||||||||||
| Contractual Obligations | ||||||||||||||||||||
| NIH milestones(1) | 1,634,125 | 1,634,125 | - | - | - | |||||||||||||||
| Leiden University milestones(2) | 323,504 | 40,438 | 283,066 | - | - | |||||||||||||||
| Car lease obligations | 304,413 | 154,351 | 150,062 | - | - | |||||||||||||||
| Total | 2,262,042 | 1,828,914 | 433,128 | - | - | |||||||||||||||
| (1) | Includes $425,000 in milestone payments. |
| (2) | Includes a €10,000 annual royalty and €50,000 upon the initiation of a Phase I study. We will update our milestone payment obligations upon releasing the Phase I data from such study. As such, the obligations above do not include a potential milestone payment of €100,000 upon the initiation of a Phase II study, €200,000 upon the initiation of a Phase III study or €500,000 upon marketing approval by any regulatory authority. |
ITEM 6. Directors, Senior Management and Employees
A. Directors and Senior Management.
The following table sets forth the members of our senior management and Board of Directors:
| Member | Position | Age | ||
| Ilan Cohn, Ph.D. | Chairman of the Board | 61 | ||
| Pnina Fishman, Ph.D. | Chief Executive Officer, Director | 68 | ||
| Motti Farbstein | Chief Operating and Financial Officer | 53 | ||
| Guy Regev | Director, Audit Committee and Compensation Committee member | 48 | ||
| Abraham Sartani, M.D. | Director | 71 | ||
| Gil Oren | Director, Audit Committee and Compensation Committee member | 64 | ||
| Israel Shamay | Director, Audit Committee and Compensation Committee member | 53 |
Ilan Cohn, Ph.D. Ilan Cohn, Ph.D. is a patent attorney and senior partner at the patent attorney firm Reinhold Cohn and Partners, where he has been an attorney since 1986. Dr. Cohn co-founded Can-Fite, served as its Chief Executive Officer until September 2004, served on our Board of Directors since 1994 and since May 30, 2013 servesas the Chairman of the Can-Fite Board of Directors. Dr. Cohn has also been a director of OphthaliX since November 21, 2011. Dr. Cohn holds a Ph.D. in biology and is a patent attorney with many years of experience in the biopharmaceutical field. He has served on the Board of Directors of a number of life science companies, including Discovery Laboratories Inc. (formerly Ansan Pharmaceuticals), a U.S. public company. Dr. Cohn has also been involved in the past in management of venture capital funds focused on investments in the life sciences industry. Dr. Cohn served a number of years as a co-chairman of the Biotech Committee of the US-Israeli Science and Technology Commission. Dr. Cohn is also currently a member of the Board of Directors of I.C.R.C Management Ltd, Famillion BVI Ltd. and Famillion Ltd. (a subsidiary of Famillion BVI Ltd.). Dr. Cohn holds a Ph.D. in Biology from the Hebrew University of Jerusalem.
| 79 |
Pnina Fishman, Ph.D. Pnina Fishman, Ph.D. co-founded Can-Fite and has served as our Chief Executive Officer and served on our Board of Directors since September 2005. She has also served as the Chief Executive Officer of OphthaliX from November 21, 2011 through December 31, 2012. Dr. Fishman is the scientific founder of Can-Fite and was previously a professor of Life Sciences and headed the Laboratory of Clinical and Tumor Immunology at the Felsenstein Medical Research Institute, Rabin Medical Center, Israel. Dr. Fishman has authored or co-authored over 150 publications and presented the findings of her research at many major scientific meetings. Her past managerial experience included seven years as Chief Executive Officer of Mor Research Application, the technology transfer arm of Clalit Health Services, the largest healthcare provider in Israel. Mor Research Application was also the first clinical research organization in Israel. Dr. Fishman currently also serves as a member of the Board of Directors of F.D Consulting Ltd., Ultratrend Ltd., EyeFite Ltd. and OphthaliX Inc. Dr. Fishman holds a Ph.D. in Immunology from the Bar Ilan University in Ramat Gan, Israel.
Motti Farbstein. Motti Farbstein has been with Can-Fite since 2003. Mr. Farbstein served as our Chief Operating Officer from August 2003 until May 2005 and from that date onwards he served as Chief Operating and Financial Officer. Mr. Farbstein also serves as a director of EyeFite Ltd. since July 2011. Mr. Farbstein’s past managerial experience includes seven years as Vice President of Mor Research Application, a company that managed the commercialization of the intellectual property of all hospitals and research centers affiliated with Clalit Health Services, which is the largest healthcare provider in Israel and was Israel’s first clinical CRO. Mr. Farbstein also has extensive experience in the data management of clinical trials.
Guy Regev. Guy Regev has over twelve years of experience in accounting, financial management and control and general management of commercial enterprises. He has served on our Board of Directors since July 2011 and has served as a member of our Audit Committee and Compensation Committee since February 2014. Mr. Regev has also been a director of OphthaliX since November 2011. Mr. Regev is currently the Chief Executive Officer of Gaon Holdings Ltd, a publicly traded Israeli holding company traded on the TASE which focuses on three areas of operation - Cleantech / Water, Financial Services, Retail/Trading. Mr. Regev is currently also the Chief Executive Officer of Middle East Tube Company Ltd a publicly traded Israeli company traded on the TASE which focuses on steel pipe manufacturing and galvanization services. Mr. Regev was the Chief Executive Officer of Shaked Global Group Ltd, a privately-held equity investment firm that provides value added capital to environmental-related companies and technologies. Prior to joining Shaked, from 2001 to 2008, Mr. Regev was Vice President of Commercial Business at Housing & Construction Holding, or HCH, Israel’s largest infrastructure company. His duties included being responsible for the consolidation and financial recovery of various business units within HCH. Prior to that, Mr. Regev carried several roles within the group including as a Chief Financial Officer and later the Chief Executive Officer of Blue-Green Ltd., the environmental services subsidiary of HCH. Between 1999 and 2001, Mr. Regev was a manager at Deloitte & Touche, Israel. Mr. Regev holds an LLB degree in Law (Israel) and is a licensed attorney and has been a licensed CPA since 1999. Mr. Regev is also a director of, The Green Way Ltd, Shtang Construction and Engineering Ltd, R.I.B.E. Consulting & Investment Ltd., Middle East Tube Company Ltd, Middle East Tube - Industries 2001 Ltd, Middle East Tubes - Galvanizing (1994) Ltd, I-Solar Greentech Ltd, Plassim Infrastructure Ltd, Plassim Advanced Solutions in Sanitation Ltd, Hakohav Valves Industries Metal (1987) Ltd, Metzerplas Agriculture Cooperative Ltd, B. Gaon Retail & Trading Ltd, Gaon Agro - Rimon Management Services Ltd, B. Gaon Business (2004) Ltd, Gaon Antan Investments Ltd, Or Asaf Investments Ltd, Hamashbir Holdings (1999) Ltd, and AHAVA Holdings LTD.
Abraham Sartani, M.D. Abraham Sartani has served on our Board of Directors since 2001. Dr. Sartani has over 30 years of experience in the pharmaceuticals industry and currently acts as a consultant to pharmaceutical and medical device companies. Dr. Sartani is a member of a number of scientific and management societies and the author or co-author of numerous publications and patents in the urology, pain treatment and hypertension fields. Dr. Sartani also currently serves on the Board of Directors of Akkadeas Pharma Srl and is a co-founding partner. From 1985 until 2008, Dr. Sartani was the Vice-President of R&D and Licensing of Recordati, a European specialty pharmaceutical company. Prior to joining Recordati, from 1980 until 1985, Dr. Sartani was employed at Farmitalia-Carlo Erba, serving in a number of capacities, including as the Medical Director for Europe.
| 80 |
Gil Oren. Gil Oren has served as external director on our Board of Directors since July 2008 and chairs both the Audit Committee and Compensation Committee since July 2008. Mr. Oren is the founder of a private consulting firm he started in 2008. Mr. Oren has over 25 years of experience in top managerial positions in various public companies in Israel and the United States and currently serves on the Board of Directors of Pointer Telocation Ltd. (NASDAQ: PNTR). From 1976 to 1992, Mr. Oren served in various positions within the Tadiran Group, including serving for five years as the Chief Financial Officer of Tadiran Electronic’s U.S. subsidiary. After serving in such capacity, Mr. Oren returned to Israel and joined Cargal, first as Vice President of Finance and then as Chief Executive Officer and General Manager. From 2002 to 2007, Mr. Oren joined SFK, a leading Israeli investment group, and served in various capacities in its portfolio companies, including as the deputy chief executive office of Urdan Industries, the chief executive officer of Itong Industries and the chairman of the Board of Directors of Orlite Industries. Mr. Oren has also served, on behalf of SFK, on the Board of Directors of various other public and private companies, including Nirlat, Aloni and Scope. Mr. Oren holds a B.A in accounting and economics from Tel Aviv University and a M.B.A from Tel Aviv University. Mr. Oren is also Certified Public Accountant.
Israel Shamay has served as external director on our Board of Directors since December 2014 and serves as a member on both the Audit Committee and Compensation Committee. Since 2012 Mr. Shamay has served as Executive Director, Strategic Initiatives and Head of the Americas Operations of MATIMOP (Israeli Industry Center for R&D), the International Operations agency of the Israeli Office of the Chief Scientist, focusing on developing and implementing cooperation platforms for industrial R&D and innovation projects in the Americas region. From 2006 until 2012 Mr. Shamay served as Executive Director of European Cooperations at MATIMOP, where he was in charge of architecting, realizing and evaluating industrial innovation cooperation frameworks at bilateral and European level, making them a major R&D cooperation instrument for Israeli industry with Europe. Between 2010 and 2011, Mr. Shamay was Head of the Israeli EUREKA Chairmanship Program (EUREKA is Europe's largest innovation network with nearly 40 member states). The Israeli EUREKA Chairmanship focused on developing new financial instruments for innovative small and medium sized enterprises and on expanding EUREKA's international dimension. From 2002 Mr. Shamay served as Israel's National Representative in several international R&D programs, from 2005 as an expert evaluator for the EU Framework Programs for R&D and from 2006 until 2009 managed the Israeli R&D collaboration with the EU Global Satellite Navigation Program – GALILEO. From 1991 till 2001 Mr. Shamay served in senior technical, marketing and executive positions in Israeli hi-tech companies operating globally, including the RAD group and Comverse Technologies. Mr. Shamay is an MBA graduate of the Recanati School of Business at the Tel-Aviv University and a graduate of the Technion in Haifa, faculty of Information Systems Engineering.
B. Compensation.
Compensation of Directors and Senior Management
The following table presents in the aggregate all compensation we paid to all of our directors and senior management as a group for the year ended December 31, 2016. It does not include any business travel, relocation, professional, and business association dues and expenses reimbursed to office holders, and other benefits commonly reimbursed or paid by companies in Israel.
| Salaries, fees, commissions, bonuses and options (thousand NIS) | ||||
| All directors and senior management as a group, consisting of 10 persons | 3,838 | |||
| 81 |
The following table presents information regarding compensation reflected in our financial statements for five most highly compensated office holders, as of December 31, 2016.
| Name and Position | Salary | Bonus | Value of Options Granted(4) | Other(5) | Total | |||||||||||||
| (NIS in thousands) | ||||||||||||||||||
| Pnina Fishman Chief Executive Officer | 1,238 | (1) | 240 | 560 | 48 | 2,086 | ||||||||||||
| Motti Farbstein Chief Financial Officer | 937 | (2) | 148 | 99 | 55 | 1,239 | ||||||||||||
| Gil Oren Director | 129 | (3) | - | 2 | - | 131 | ||||||||||||
| Guy Regev External Director | 120 | (3) | - | - | - | 120 | ||||||||||||
| Israel Shamay External Director | 128 | (3) | - | - | - | 128 | ||||||||||||
(1)
|
Amount represents consulting fee. |
| (2) | Salary includes Mr. Farbstein’s gross salary plus payment of social benefits made by us on behalf of such person. Such benefits may include, to the extent applicable, payments, contributions and/or allocations for savings funds (e.g., managers’ life insurance policy), education funds (referred to in Hebrew as “keren hishtalmut”), pension, severance, risk insurances (e.g., life, or work disability insurance), payments for social security payments and tax gross-up payments, vacation, medical insurance and benefits, convalescence or recreation pay and other benefits and perquisites consistent with our policies. |
| (3) | Amount represents fees for board service. |
| (4) | The value of options is the expense recorded in our financial statements for the period ended December 31, 2016 with respect to all options granted to such person. |
| (5) | Amount represents cost of use of company car. |
The following table sets forth information with respect to the options granted to the members of our executive officers and directors for the year ended December 31, 2016.
| Name | Date of Grant | Purchase Price (in NIS) | Number of Options | Vesting Period | Expiration Date | Total Benefit (in NIS) | Benefit recognized in 2016 (in NIS) | |||||||||||||||
| Motti Farbstein | 17/2/16 | 4.317 | 60,000 | 4 years | 17/2/26 | 159,644 | 80,533 | |||||||||||||||
Each director other than our Chief Executive Officer and Avraham Sartani, is entitled to the payment of annual fee of NIS 48,721 (approximately $12,671), and payment of NIS 3,256 (approximately $847) per meeting for participating in meetings of the board and committees of the board. The annual fee shall not exceed the annual fee of an expert external director set forth in the Companies Regulations (Rules regarding Compensation and Expenses of External Directors) 5760-2000 as adjusted by the Companies Regulations (Relief for Public Companies with Shares Listed for Trading on a Stock Market Outside of Israel), 5760-2000. The compensation awarded for participating in resolutions that are adopted without an actual convening (i.e., unanimous written resolutions) and for participating through telephone meetings will be reduced as follows: (1) for resolutions that will be adopted without an actual convening, the participation compensation will be reduced by 50%; and (2) for participation through telephone meetings, the participation compensation will be reduced by 40%. The participation compensation and the annual fee is inclusive of all expenses incurred by our directors in connection with their participation in a meeting held at our offices or with regard to resolutions resolved by written consent or teleconference. Avraham Sartani is entitled to a fee of $1,000 per meeting. In addition, our directors (other than our Chief Executive Officer and external directors) are entitled to reimbursement for expenses related to their participation at meetings taking place not at our offices and outside their respective residency area.
| 82 |
Employment and Consulting Agreements
We have entered into employment or consulting agreements with our directors, senior management and key service providers. All of these agreements contain customary provisions regarding noncompetition, confidentiality of information and assignment of proprietary information and inventions. However, the enforceability of the noncompetition provisions may be limited under applicable law.
The following are summary descriptions of certain agreements to which we are a party. The descriptions provided below do not purport to be complete and are qualified in their entirety by the complete agreements, which are attached as exhibits to this Annual Report on Form 20-F.
Service Management Agreement with F.D. Consulting: On June 27, 2002, we entered into a Service Management Agreement with F.D. Consulting, a company partially owned by Pnina Fishman, pursuant to which Dr. Fishman began serving as our Chief Scientific Officer and later became our Chief Executive Officer and is a member of our Board of Directors and continues to be retained through this agreement. F.D. Consulting’s current gross monthly fee is NIS 103,200 which is linked to the Israeli CPI and fluctuates accordingly. Dr. Fishman, through F.D. Consulting, is also entitled to reimbursement for reasonable out-of-pocket expenses and use of a company automobile and mobile phone.
The term of F.D. Consulting’s service management agreement is indefinite, unless earlier terminated for cause by us or without cause by either party, subject to three months’ advanced notice.
Dr. Fishman is also entitled to receive options exercisable into our ordinary shares from time to time. As of March 29, 2017, we have granted her options to purchase an aggregate of 744,443 ordinary shares, of which (i) 241,613 were exercised into ordinary shares, (ii) options to purchase 195,630 ordinary shares expired (iii) 2,680,000 options to purchase 107,200 ordinary shares have an exercise price of NIS 0.644 per option, are fully vested and expire on January 13, 2021, and (iii) 200,000 options to purchase 200,000 ordinary shares have an exercise price of NIS 3.573 per ordinary share, vesting on a quarterly basis over three years commencing October 22, 2015, and expire on October 22, 2025.
Employment and Non-Competition Agreement with Motti Farbstein: On September 1, 2003 we entered into an employment and non-competition agreement with Motti Farbstein pursuant to which Mr. Farbstein began serving as our Director of Clinical Operations and Administrative Affairs on September 1, 2003 and is currently serving as our Chief Operating and Financial Officer. Mr. Farbstein’s current gross monthly salary is NIS 49,450. Mr. Farbstein is entitled to an allocation to a manager’s insurance policy equivalent to an amount up to 13-1/3% of his gross monthly salary, up to 2-1/2% of his gross monthly salary for disability insurance and 7-1/2% of his gross monthly salary for a study fund. The foregoing amounts are paid by us. Five percent of his gross monthly salary is deducted for the manager’s insurance policy and 2-1/2% is deducted for the study fund. Mr. Farbstein is also entitled to reimbursement for reasonable out-of-pocket expenses, including travel expenses, and use of a company automobile and mobile phone.
The term of Mr. Farbstein’s employment and non-competition agreement is indefinite, unless earlier terminated for just cause by either party, upon the death, disability or retirement age, or without cause by either party, subject to 60 days’ advanced notice.
Mr. Farbstein is also entitled to receive options exercisable into our ordinary shares from time to time. As of March 29, 2017, we have granted him options to purchase an aggregate of 114,195 ordinary shares, of which (i) options to purchase ordinary shares were exercised into 1,133 ordinary shares, (ii) options to purchase 12,887 ordinary shares expired, (iii) 554,387 options are exercisable into 22,175 ordinary shares at an exercise price of NIS 0.307 per option, are fully vested, and expire on November 26, 2018, (iii) 100,000 options are exercisable into 4,000 ordinary shares at an exercise price of NIS 0.385 per option, are fully vested, and expire on May 2, 2022, (iv) 100,000 options are exercisable into 4,000 ordinary shares at an exercise price of NIS 0.326 per option are fully vested, and expire on March 20, 2023, (v) 10,000 options to purchase 10,000 ordinary shares at an exercise price of NIS 8.1205 per option, vesting on a quarterly basis over four years commencing March 19, 2015, and expire on March 18, 2025, (vi) 60,000 options to purchase 60,000 ordinary shares at an exercise price of NIS 4.317 per ordinary shares, vesting on a quarterly basis over four years commencing February 18, 2016 and expire on February 18, 2026.
Consulting Agreement with BioStrategics: On September 27, 2005, we entered into a consulting agreement with BioStrategics through its President, Michael Silverman pursuant to which Dr. Silverman began serving as our Medical Director. Dr. Silverman has extensive experience in clinical development acquired through his involvement in clinical development in large pharmaceutical and small biopharmaceutical companies. He was involved in international clinical research, market-oriented strategic planning, and the challenges of managing research and development portfolios in various capacities at Sterling Winthrop Research Institute and subsequently at Sandoz Research Institute.
| 83 |
BioStrategics’ current fee is $325 per hour with a maximum daily fee of $2,600. In addition, BioStrategics is entitled to reimbursement for reasonable pre-approved expenses. The term of the consulting agreement is currently on a year-to-year basis, unless earlier terminated by either party upon 30 days’ prior written notice or immediately by either party if such termination is for cause.
Master Services Agreement with Accellient Partners: On May 10, 2010, we entered into a Master Services Agreement with Accellient Partners, a company owned by William Kerns, who currently serves as our current Vice President of Drug Development. Dr. Kerns has over 20 years of experience in Pharmaceutical Research and Development at SmithKline Beecham and Eisai Pharmaceuticals. As a Senior Executive he has participated in the development of drugs for over 100 Phase I studies and 13 NDA’s and/or Marketing Authorization Applications. Dr. Kerns has chaired a FDA committee on biomarkers and he is an expert in preclinical development and regulatory strategy.
According to the agreement, consulting services are provided by Accellient Partners’ personnel in accordance with individual work orders that are executed from time to time. Each individual work order defines the scope of work to be provided and sets forth the fees to be paid to Accellient Partners.
Beginning on May 10, 2012, the term of the master services agreement is on a month-to-month basis, unless terminated by us upon 30 days’ prior written notice, by us at any time if Accellient Partners commits a breach and fails to cure, or by Accellient Partners upon 30 days’ prior written notice if we commit a breach and fail to cure.
Reinhold Cohn and Partners: Reinhold Cohn and Partners, an Israeli partnership, of which Ilan Cohn, Ph.D. is a partner provides intellectual property services to us in the ordinary course of business.
C. Board Practices
General
According to the Israeli Companies Law, the management of our business is vested in our Board of Directors. Our Board of Directors may exercise all powers and may take all actions that are not specifically granted to our shareholders. Our executive officers are responsible for our day-to-day management and have individual responsibilities established by our Board of Directors. Executive officers are appointed by and serve at the discretion of our Board of Directors, subject to any applicable employment agreements we have entered into with the executive officers. See “Item 6—Directors, Senior Management and Employees—Compensation—Employment and Consulting Agreements.”
Election of Directors and Terms of Office
Our Board of Directors currently consists of six members. Other than our two external directors, our directors are elected by an ordinary resolution at the annual general meeting of our shareholders. The nomination of our directors is proposed by the Board of Directors. Our board has the authority to add additional directors up to the maximum number of 12 directors allowed under our Articles. Such directors appointed by the board serve until the next annual general meeting of the shareholders. Unless they resign before the end of their term or are removed in accordance with our Articles of Association, all of our directors, other than our external directors, will serve as directors until our next annual general meeting of shareholders. On January 3, 2017, at an annual general meeting of our shareholders, Pnina Fishman, Ilan Cohn, Avi Sartani, and Guy Regev were re-elected to serve as directors for a term expiring at our next annual general meeting of shareholders and until his or her respective successor is duly elected. On July 14, 2014, at an annual general meeting of our shareholders Gil Oren was re-elected to serve as one of our external directors for an additional three-year term ending July 9, 2017. On December 31, 2014, at a special meeting of our shareholders, Israel Shamay was elected to serve for a three-year term ending December 30, 2017 as one of our external directors. Israel Shamay may be re-elected for another two three-year terms. Gil Oren may not be re-elected to serve as an external director as he was elected for three terms, the maximum term according to the provisions of the Israeli Companies Law. On May 30, 2013, Ilan Cohn was appointed as Chairman of the Board.
None of our directors or officers has any family relationship with any other director or officer. None of our directors have service contracts that provide for benefits upon termination of his or her directorship with us, other than the payment of salary due, accrued and unpaid as of and through the date of termination. See “Item 6—Directors, Senior Management and Employees—Compensation—Employment and Consulting Agreements.”
| 84 |
Chairman of the Board. Under the Israeli Companies Law, without shareholder approval, a person cannot hold the role of both chairman of the board of directors and chief executive officer of a company. Furthermore, a person who is directly or indirectly subordinate to a chief executive officer of a company may not serve as the chairman of the board of directors of that company and the chairman of the board of directors may not otherwise serve in any other capacity in a company or in a subsidiary of that company other than as the chairman of the board of directors of such a subsidiary.
The Israeli Companies Law provides that an Israeli company may, under certain circumstances, exculpate an office holder from liability with respect to a breach of his duty of care toward the company if appropriate provisions allowing such exculpation are included in its articles of association. Our Articles of Association permit us to maintain directors’ and officers’ liability insurance and to indemnify our directors and officers for actions performed on behalf of us, subject to specified limitations. We maintain a directors and officers insurance policy which covers the liability of our directors and officers as allowed under the Israeli Companies Law.
The term office holder is defined in the Israeli Companies Law as a director, general manager, chief business manager, deputy general manager, vice general manager, executive vice president, vice president, any other manager directly subordinate to the general manager or any other person assuming the responsibilities of any of the foregoing positions, without regard to such person’s title.
External and Independent Directors
Under the Israeli Companies Law, the boards of directors of companies whose shares are publicly traded, either within or outside of Israel, are required to include at least two members who qualify as external directors.
External directors must be elected by a majority vote of the shares present and voting at a shareholders meeting, provided that either:
| ● | the majority of the shares that are voted at the meeting, including at least a majority of the shares held by non-controlling shareholders who do not have a personal interest in the election of the external director (other than a personal interest not deriving from a relationship with a controlling shareholder) who voted at the meeting, excluding abstentions, vote in favor of the election of the external director; or |
| ● | the total number of shares held by non-controlling, disinterested shareholders (as described in the preceding bullet point) that are voted against the election of the external director does not exceed 2% of the aggregate voting rights in the company. |
The term controlling shareholder is defined in the Israeli Companies Law as a shareholder with the ability to direct the activities of the Company, other than by virtue of being an office holder, but there is a presumption that a shareholder holding 25% of the shares of the Company is regarded as a controlling shareholder. A person may not serve as an external director of a company if (i) such person is a relative of a controlling shareholder of a company or (ii) at the date of such person’s appointment or within the prior two years, such person, such person’s relative, partner, employer or any entity under such person’s control or anyone to whom such person is subordinate, whether directly or indirectly, has or had any affiliation with (a) the company, (b) the controlling shareholder at the time of such person’s appointment or (c) any entity that is either controlled by the company or under common control with the company at the time of such appointment or during the prior two years. If a company does not have a controlling shareholder or a shareholder who holds company shares entitling him to vote at least 25% of the votes in a shareholders meeting, then a person may not serve as an external director if, such person or such person’s relative, partner, employer or any entity under such person’s control, has or had, on or within the two years preceding the date of the person’s appointment to serve as an external director, any affiliation with the chairman of our board of directors, chief executive officer, a substantial shareholder who holds at least 5% of the issued and outstanding shares of the company or voting rights which entitle him to vote at least 5% of the votes in a shareholders meeting, or the chief financial officer of the company.
The term affiliation includes:
| ● | an employment relationship; |
| ● | a business or professional relationship even if not maintained on a regular basis (excluding insignificant relationships); |
| ● | control; and |
| ● | service as an office holder, excluding service as a director in a private company prior to the first offering of its shares to the public if such director was appointed as a director of the private company in order to serve as an external director following the public offering. |
| 85 |
The term relative is defined as a spouse, sibling, parent, grandparent or descendant; a spouse’s sibling, parent or descendant; and the spouse of each of the foregoing persons.
In addition, no person may serve as an external director if that person’s professional activities create, or may create, a conflict of interest with that person’s responsibilities as a director or otherwise interfere with that person’s ability to serve as an external director or if the person is an employee of the Israel Securities Authority, or ISA, or of the TASE. Furthermore, a person may not continue to serve as an external director if he or she received direct or indirect compensation from the company for his or her role as a director. This prohibition does not apply to compensation paid or given in accordance with regulations promulgated under the Israeli Companies Law or amounts paid pursuant to indemnification and/or exculpation contracts or commitments and insurance coverage. If, at the time an external director is appointed, all current members of the board of directors not otherwise affiliated with the company are of the same gender, then that external director must be of the other gender. In addition, a director of a company may not be elected as an external director of another company if, at that time, a director of the other company is acting as an external director of the first company.
Following the termination of an external director’s service on a board of directors, such former external director and his or her spouse and children may not be provided with a direct or indirect benefit by the company, its controlling shareholder or any entity under its controlling shareholder’s control. This includes engagement to serve as an executive officer or director of the company or a company controlled by its controlling shareholder, or employment by, or providing services to, any such company for consideration, either directly or indirectly, including through a corporation controlled by the former external director, for a period of two years (and for a period of one year with respect to relatives of the former external director).
The Israeli Companies Law provides that an external director must meet certain professional qualifications or have financial and accounting expertise and that at least one external director must have financial and accounting expertise. However, if at least one of our other directors (i) meets the independence requirements of the Securities Exchange Act of 1934, as amended, (ii) meets the standards of the NYSE MKT rules for membership on the audit committee and (iii) has financial and accounting expertise as defined in the Israeli Companies Law and applicable regulations, then neither of our external directors is required to possess financial and accounting expertise as long as both possess other requisite professional qualifications. Our Board of Directors is required to determine whether a director possesses financial and accounting expertise by examining whether, due to the director’s education, experience and qualifications, the director is highly proficient and knowledgeable with regard to business-accounting issues and financial statements, to the extent that the director is able to engage in a discussion concerning the presentation of financial information in our financial statements, among others. The regulations define a director with the requisite professional qualifications as a director who satisfies one of the following requirements: (i) the director holds an academic degree in either economics, business administration, accounting, law or public administration; (ii) the director either holds an academic degree in any other field or has completed another form of higher education in our primary field of business or in an area which is relevant to the office of an external director; or (iii) the director has at least five years of experience serving in any one of the following, or at least five years of cumulative experience serving in two or more of the following capacities: (a) a senior business management position in a corporation with a substantial scope of business; (b) a senior position in our primary field of business; or (c) a senior position in public administration. Gil Oren, who is one of our external directors, meets the required qualifications and has financial and accounting expertise as required by the Israeli Companies Law, while Guy Regev, and independent director, also meets the required qualifications and has financial and accounting expertise as required by the Israeli Companies Law.
The Israeli Companies Law defines an independent director as a director who complies with the following and was appointed as such in accordance with Chapter 1 of Part 56 of the Israeli Companies Law: (1) the director complies with the qualification to serve as an external director as set out in Sections 240 (b)-(f) of the Israeli Companies Law and the audit committee has approved such compliance; and (2) the director has not served as a director of the company for more than nine consecutive years (which, for such purpose, does not include breaks in such service for periods of less than two year).
If an external directorship becomes vacant and there are less than two external directors on the board of directors at the time, then the board of directors is required under the Israeli Companies Law to call a shareholders' meeting as soon as possible to appoint a replacement external director.
| 86 |
Each committee of the board of directors that is authorized to exercise the powers of the board of directors must include at least one external director, except that the audit committee and compensation committee must each include all external directors then serving on the board of directors. Under the Israeli Companies Law, external directors of a company are prohibited from receiving, directly or indirectly, any compensation for their services as external directors, other than compensation and reimbursement of expenses pursuant to applicable regulations promulgated under the Israeli Companies Law. Compensation of an external director is determined prior to his or her appointment and may not be changed during his or her term subject to certain exceptions.
Israel Shamay and Gil Oren serve as external directors on our Board of Directors pursuant to the provisions of the Israeli Companies Law. They both serve on our audit committee and our compensation committee. Our Board of Directors has determined that Gil Oren possesses accounting and financial expertise, and that both of our external directors possess the requisite professional qualifications. In addition to our external directors, Guy Regev and Avi Sartani serve as independent directors on our Board of Directors. Guy Regev also serves on our audit committee and our compensation committee.
Audit Committee
The Israeli Companies Law requires public companies to appoint an audit committee. The responsibilities of the audit committee include identifying irregularities in the management of our business and approving related party transactions as required by law. An audit committee must consist of at least three directors, including all of its external directors and a majority of independent directors. The chairman of the board of directors, any director employed by or otherwise providing services to the company, and a controlling shareholder or any relative of a controlling shareholder, may not be a member of the audit committee. An audit committee may not approve an action or a transaction with a controlling shareholder, or with an office holder, unless at the time of approval two external directors are serving as members of the audit committee and at least one of the external directors was present at the meeting in which an approval was granted.
Our audit committee is currently comprised of three independent non-executive directors. The audit committee is chaired by Gil Oren, who serves as the audit committee financial expert, with Israel Shamay and Guy Regev as members. Our audit committee meets at least four times a year and monitors the adequacy of our internal controls, accounting policies and financial reporting. It regularly reviews the results of the ongoing risk self-assessment process, which we undertake, and our interim and annual reports prior to their submission for approval by the full Board of Directors. The audit committee oversees the activities of the internal auditor, sets its annual tasks and goals and reviews its reports. The audit committee reviews the objectivity and independence of the external auditors and also considers the scope of their work and fees.
Our audit committee provides assistance to our Board of Directors in fulfilling its legal and fiduciary obligations in matters involving our accounting, auditing, financial reporting, internal control and legal compliance functions by pre-approving the services performed by our independent accountants and reviewing their reports regarding our accounting practices and systems of internal control over financial reporting. Our audit committee also oversees the audit efforts of our independent accountants and takes those actions that it deems necessary to satisfy itself that the accountants are independent of management.
Under the Israeli Companies Law, our audit committee is responsible for (i) determining whether there are deficiencies in the business management practices of our company, including in consultation with our internal auditor or the independent auditor, and making recommendations to the Board of Directors to improve such practices and amend such deficiencies, (ii) determining whether certain related party transactions (including transactions in which an office holder has a personal interest) should be deemed as material or extraordinary, and to approve such transactions (which may be approved according to certain criteria set out by our audit committee on an annual basis) (see "—Approval of related party transactions under Israeli Law"), (iii) to establish procedures to be followed in respect of related party transactions with a controlling shareholder (where such are not extraordinary transactions), which may include, where applicable, the establishment of a competitive process for such transaction, under the supervision of the audit committee, or individual, or other committee or body selected by the audit committee, in accordance with criteria determined by the audit committee; (iv) to determine procedures for approving certain related party transactions with a controlling shareholder, which having been determined by the audit committee not to be extraordinary transactions, were also determined by the audit committee not to be negligible transactions; (v) approves the working plan of the internal auditor, to examine such working plan before its submission to the Board and propose amendments thereto, (vi) examining our internal controls and internal auditor's performance, including whether the internal auditor has sufficient resources and tools to dispose of its responsibilities, (vii) examining the scope of our auditor's work and compensation and submitting a recommendation with respect thereto to our Board of Directors or shareholders, depending on which of them is considering the appointment of our auditor, and (viii) establishing procedures for the handling of employees' complaints as to the management of our business and the protection to be provided to such employees.
| 87 |
We have adopted a written charter for our audit committee, setting forth its responsibilities as outlined by the regulations of the SEC. In addition, our audit committee has adopted procedures for the receipt, retention and treatment of complaints we may receive regarding accounting, internal accounting controls or auditing matters and the submission by our employees of concerns regarding questionable accounting or auditing matters. In addition, SEC rules mandate that the audit committee of a listed issuer consist of at least three members, all of whom must be independent, as such term is defined by rules and regulations promulgated by the SEC. We are in compliance with the independence requirements of the SEC rules.
Any person who is not eligible to serve on the audit committee is further restricted from participating in its meetings and votes, unless the chairman of the audit committee determines that such person’s presence is necessary in order to present a certain matter; provided, however, that company employees who are not controlling shareholders or relatives of such shareholders may be present in the meetings, but not for actual voting, and likewise, company counsel and secretary who are not controlling shareholders or relatives of such shareholders may be present in the meetings and for actual voting if such presence is requested by the audit committee.
In addition to the above, all such committee’s members must apply with the following requirements:
| ● | All members shall be members of the board of directors of the company. |
| ● | At least one of the committee’s members shall have financial and accounting expertise and the rest of the committee’s members must have the ability to read and understand financial statements. |
Our company, through our audit committee, is in full compliance with the above requirements.
Financial Statement Examination Committee
Under the Israeli Companies Law, the board of directors of a public company must appoint a financial statement examination committee, which consists of members with accounting and financial expertise or the ability to read and understand financial statements. According to a resolution of our Board of Directors, the audit committee has been assigned the responsibilities and duties of a financial statements examination committee, as permitted under relevant regulations promulgated under the Israeli Companies Law. From time to time as necessary and required to approve our financial statements, the audit committee holds separate meetings, prior to the scheduled meetings of the entire Board of Directors regarding financial statement approval. The function of a financial statements examination committee is to discuss and provide recommendations to its board of directors (including the report of any deficiency found) with respect to the following issues: (i) estimations and assessments made in connection with the preparation of financial statements; (ii) internal controls related to the financial statements; (iii) completeness and propriety of the disclosure in the financial statements; (iv) the accounting policies adopted and the accounting treatments implemented in material matters of the company; (v) value evaluations, including the assumptions and assessments on which evaluations are based and the supporting data in the financial statements. Our independent auditors and our internal auditors are invited to attend all meetings of audit committee when it is acting in the role of the financial statements examination committee.
Compensation Committee
Amendment no. 20 to the Companies Law was published on November 12, 2012 and became effective on December 12, 2012, or Amendment no. 20.In general, Amendment no. 20 requires public companies to appoint a compensation committee and to adopt a compensation policy with respect to its officers, or the Compensation Policy. In addition, Amendment no. 20 addresses the corporate approval process required for a public company's engagement with its officers (with specific reference to a director, a non-director officer, a chief executive officer and controlling shareholders and their relatives who are employed by the company).
The compensation committee shall be nominated by the board of directors and be comprised of its members. The compensation committee must consist of at least three members. All of the external directors must serve on the compensation committee and constitute a majority of its members. The remaining members of the compensation committee must be directors who qualify to serve as members of the audit committee (including the fact that they are independent) and their compensation should be identical to the compensation paid to the external directors of the company.
| 88 |
Similar to the rules that apply to the audit committee, the compensation committee may not include the chairman of the board, or any director employed by the Company, by a controlling shareholder or by any entity controlled by a controlling shareholder, or any director providing services to the company, to a controlling shareholder or to any entity controlled by a controlling shareholder on a regular basis, or any director whose primary income is dependent on a controlling shareholder, and may not include a controlling shareholder or any of its relatives. Individuals who are not permitted to be compensation committee members may not participate in the committee’s meetings other than to present a particular issue; provided, however, that an employee that is not a controlling shareholder or relative may participate in the committee’s discussions, but not in any vote, and our legal counsel and corporate secretary may participate in the committee’s discussions and votes if requested by the committee.
The roles of the compensation committee are, among others, to: (i) recommend to the board of directors the Compensation Policy for office holders and recommend to the board once every three years the extension of a Compensation Policy that had been approved for a period of more than three years; (ii) recommend to the directors any update of the Compensation Policy, from time to time, and examine its implementation; (iii) decide whether to approve the terms of office and of employment of office holders that require approval of the compensation committee; and (iv) decide, in certain circumstances, whether to exempt the approval of terms of office of a chief executive officer from the requirement of shareholder approval.
The compensation policy requires the approval of the general meeting of shareholders with a “Special Majority”, which requires a majority of the shareholders of the company who are not either a controlling shareholder or an "interested party" in the proposed resolution, or that shareholders holding less than 2% of the voting power in the company voted against the proposed resolution at such meeting. However, under special circumstances, the board of directors may approve the compensation policy without shareholder approval, if the compensation committee and thereafter the board of directors decided, based on substantiated reasons after they have reviewed the compensation policy again, that the compensation policy is in the best interest of the company. The Compensation Policy policy is required to be brought before the shareholders of the Company once every three years for approval.
Under the Israeli Companies Law, our compensation policy must generally serve as the basis for corporate approvals with respect to the financial terms of employment or engagement of office holders, including exemption, insurance, indemnification or any monetary payment or obligation of payment in respect of employment or engagement. The compensation policy must relate to certain factors, including advancement of the company's objective, the company's business plan and its long term strategy, and creation of appropriate incentives for office holders. It must also consider, among other things, the company's risk management, size and nature of its operations. The compensation policy must furthermore consider the following additional factors:
| ● | The knowledge, skills, expertise, and accomplishments of the relevant office holder; |
| ● | The office holder's roles and responsibilities and prior compensation agreements with him or her; |
| ● | The relationship between the terms offered and the average compensation of the other employees of the company, including those employed through manpower companies; |
| ● | The impact of disparities in salary upon work relationships in the company; |
| ● | The possibility of reducing variable compensation at the discretion of the board of directors; the possibility of setting a limit on the exercise value of non-cash variable equity-based compensation; and |
| ● | As to severance compensation, the period of service of the office holder, the terms of his or her compensation during such service period, the company's performance during that period of service, the person's contributions towards the company's achievement of its goals and the maximization of its profits, and the circumstances under which the person is leaving the company. | |
| The Compensation Policy must also include the following principles: | ||
| ● | the link between variable compensation and the long term performance and measurable criteria; |
| ● | the relationship between variable and fixed compensation, and the ceiling for the value of variable compensation; |
| ● | the conditions under which an office holder would be required to repay compensation paid to him or her if it was later shown that the data upon which such compensation was based was inaccurate and was required to be restated in the company's financial statements; |
| ● | the minimum holding or vesting period for variable, equity-based compensation; and |
| ● | maximum limits for severance compensation. |
| 89 |
The Compensation Policy was approved by the general meeting of shareholders on January 19, 2017 after discussions and recommendation of the compensation committee and approval by the Board of Directors. Moreover, the approval of the compensation committee is required in order to approve terms of office and/or employment of office holders.
Gil Oren is the chairman of our compensation committee. Israel Shamay and Guy Regev serve as the other members of our compensation committee.
Under Amendment no. 27 to the Israeli Companies Law, which became effective as of February 17, 2016, the audit committee of an Israeli public company which has been established and conducts itself also in accordance with provisions governing the composition of the compensation committee as set forth in the Israeli Companies Law, may act in lieu of a compensation committee with respect to the responsibilities of a compensation committee which are set forth in the Israeli Companies Law.
Approval of Related Party Transactions under the Israeli Companies Law
Fiduciary duties of the office holders
The Israeli Companies Law imposes a duty of care and a duty of loyalty on all office holders of a company. The duty of care of an office holder is based on the duty of care set forth in connection with the tort of negligence under the Israeli Torts Ordinance (New Version) 5728-1968. This duty of care requires an office holder to act with the degree of proficiency with which a reasonable office holder in the same position would have acted under the same circumstances. The duty of care includes a duty to use reasonable means, in light of the circumstances, to obtain:
| ● | information on the advisability of a given action brought for his or her approval or performed by virtue of his or her position; and |
| ● | all other important information pertaining to these action |
The duty of loyalty requires an office holder to act in good faith and for the benefit of the company, and includes the duty to:
| ● | refrain from any act involving a conflict of interest between the performance of his or her duties in the company and his or her other duties or personal affairs; |
| ● | refrain from any activity that is competitive with the business of the company; |
| ● | refrain from exploiting any business opportunity of the company for the purpose of gaining a personal advantage for himself or herself or others; and |
| ● | disclose to the company any information or documents relating to our affairs which the office holder received as a result of his or her position as an office holder. |
We may approve an act performed in breach of the duty of loyalty of an office holder provided that the office holder acted in good faith, the act or its approval does not harm the company, and the office holder discloses his or her personal interest, as described below.
| 90 |
Disclosure of personal interests of an office holder and approval of acts and transactions
The Israeli Companies Law requires that an office holder promptly disclose to the company any personal interest that he or she may have and all related material information or documents relating to any existing or proposed transaction by the company. An interested office holder’s disclosure must be made promptly and in any event no later than the first meeting of the board of directors at which the transaction is considered. An office holder is not obligated to disclose such information if the personal interest of the office holder derives solely from the personal interest of his or her relative in a transaction that is not considered as an extraordinary transaction.
The term personal interest is defined under the Israeli Companies Law to include the personal interest of a person in an action or in the business of a company, including the personal interest of such person’s relative or the interest of any corporation in which the person is an interested party, but excluding a personal interest stemming solely from the fact of holding shares in the company. A personal interest furthermore includes the personal interest of a person for whom the office holder holds a voting proxy or the interest of the office holder with respect to his or her vote on behalf of the shareholder for whom he or she holds a proxy even if such shareholder itself has no personal interest in the approval of the matter. An office holder is not, however, obliged to disclose a personal interest if it derives solely from the personal interest of his or her relative in a transaction that is not considered an extraordinary transaction.
Under the Israeli Companies Law, an extraordinary transaction which requires approval is defined as any of the following:
| ● | a transaction other than in the ordinary course of business; |
| ● | a transaction that is not on market terms; or |
| ● | a transaction that may have a material impact on our profitability, assets or liabilities. |
Under the Israeli Companies Law, once an office holder has complied with the disclosure requirement described above, a company may approve a transaction between the company and the office holder or a third party in which the office holder has a personal interest, or approve an action by the office holder that would otherwise be deemed a breach of duty of loyalty. However, a company may not approve a transaction or action that is adverse to our interest or that is not performed by the office holder in good faith.
Under the Israeli Companies Law, unless the articles of association of a company provide otherwise, a transaction with an office holder, a transaction with a third party in which the office holder has a personal interest, and an action of an office holder that would otherwise be deemed a breach of duty of loyalty requires approval by the board of directors. Our Articles of Association do not provide otherwise. If the transaction or action considered is (i) an extraordinary transaction, (ii) an action of an office holder that would otherwise be deemed a breach of duty of loyalty and may have a material impact on a company’s profitability, assets or liabilities, (iii) an undertaking to indemnify or insure an office holder who is not a director, or (iv) for matters considered an undertaking concerning the terms of compensation of an office holder who is not a director, including, an undertaking to indemnify or insure such office holder, then approval by the audit committee is required prior to approval by the board of directors. Arrangements regarding the compensation, indemnification or insurance of a director require the approval of the audit committee, board of directors and shareholders, in that order.
A director who has a personal interest in a matter that is considered at a meeting of the board of directors or the audit committee may generally not be present at the meeting or vote on the matter, unless a majority of the directors or members of the audit committee have a personal interest in the matter or the chairman of the audit committee or board of directors, as applicable, determines that he or she should be present to present the transaction that is subject to approval. If a majority of the directors have a personal interest in the matter, such matter would also require approval of the shareholders of the company.
Disclosure of personal interests of a controlling shareholder and approval of transactions
Under the Israeli Companies Law and a recent amendment thereto, the disclosure requirements that apply to an office holder also apply to a controlling shareholder of a public company. See “— Audit Committee” for a definition of controlling shareholder. Extraordinary transactions with a controlling shareholder or in which a controlling shareholder has a personal interest, including a private placement in which a controlling shareholder has a personal interest, as well as transactions for the provision of services whether directly or indirectly by a controlling shareholder or his or her relative, or a company such controlling shareholder controls, and transactions concerning the terms of engagement of a controlling shareholder or a controlling shareholder’s relative, whether as an office holder or an employee, require the approval of the audit committee, the board of directors and a majority of the shares voted by the shareholders of the company participating and voting on the matter in a shareholders’ meeting. In addition, such shareholder approval must fulfill one of the following requirements:
| ● | at least a majority of the shares held by shareholders who have no personal interest in the transaction and are voting at the meeting must be voted in favor of approving the transaction, excluding abstentions; or | |
| ● | the shares voted by shareholders who have no personal interest in the transaction who vote against the transaction represent no more than 2% of the voting rights in the company. |
| 91 |
To the extent that any such transaction with a controlling shareholder is for a period extending beyond three years, approval is required once every three years, unless the audit committee determines that the duration of the transaction is reasonable given the circumstances related thereto.
Duties of shareholders
Under the Israeli Companies Law, a shareholder has a duty to refrain from abusing its power in the company and to act in good faith and in an acceptable manner in exercising its rights and performing its obligations to the company and other shareholders, including, among other things, voting at general meetings of shareholders on the following matters:
| ● | an amendment to the articles of association; |
| ● | an increase in our authorized share capital; |
| ● | a merger; |
| ● | an increase in our authorized share capital; and |
| ● | the approval of related party transactions and acts of office holders that require shareholder approval. |
A shareholder also has a general duty to refrain from discriminating against other shareholders.
The remedies generally available upon a breach of contract will also apply to a breach of the above mentioned duties, and in the event of discrimination against other shareholders, additional remedies are available to the injured shareholder.
In addition, any controlling shareholder, any shareholder that knows that its vote can determine the outcome of a shareholder vote and any shareholder that, under a company’s articles of association, has the power to appoint or prevent the appointment of an office holder, or has another power with respect to a company, is under a duty to act with fairness towards the company. The Israeli Companies Law does not describe the substance of this duty except to state that the remedies generally available upon a breach of contract will also apply in the event of a breach of the duty to act with fairness, taking the shareholder’s position in the company into account.
Exculpation, Insurance and Indemnification of Directors and Officers
Under the Israeli Companies Law, a company may not exculpate an office holder from liability for a breach of the duty of loyalty. An Israeli company may exculpate an office holder in advance from liability to us, in whole or in part, for damages caused to us as a result of a breach of duty of care but only if a provision authorizing such exculpation is included in its articles of association. Our amended and restated articles of association include such a provision. We may not exculpate in advance a director from liability arising out of a prohibited dividend or distribution to shareholders.
| 92 |
Under the Israeli Companies Law and the Israeli Securities Law, a company may indemnify, or undertake in advance to indemnify, an office holder, provided its articles of association include a provision authorizing such indemnification, for the following liabilities and expenses imposed on an office holder or incurred by office holder due to acts performed by him or her as an office holder:
| ● | Financial liability incurred by or imposed on him or her in favor of another person pursuant to a judgment, including a settlement or arbitrator's award approved by a court. However, if an undertaking to indemnify an office holder with respect to such liability is provided in advance, then such an undertaking must be limited to events which, in the opinion of the board of directors, can be foreseen based on our activities when the undertaking to indemnify is given, and to an amount or according to criteria determined by the board of directors as reasonable under the circumstances, and such undertaking shall detail the abovementioned foreseen events and amount or criteria; |
| ● | Reasonable litigation expenses, including attorneys' fees, incurred by the office holder as a result of an investigation or proceeding instituted against him or her by an authority authorized to conduct such investigation or proceeding, provided that (i) no indictment was filed against such office holder as a result of such investigation or proceeding; and (ii) no financial liability was imposed upon him or her as a substitute for the criminal proceeding as a result of such investigation or proceeding or, if such financial liability was imposed, it was imposed with respect to an offense that does not require proof of criminal intent or as a monetary sanction; |
| ● | Reasonable litigation expenses, including attorneys' fees, incurred by the office holder or imposed by a court in proceedings instituted against him or her by us, on our behalf, or by a third party, or in connection with criminal proceedings in which the office holder was acquitted, or as a result of a conviction for an offense that does not require proof of criminal intent; and |
| ● | Expenses, including reasonable litigation expenses and legal fees, incurred by an office holder in relation to an administrative proceeding instituted against such office holder, or certain compensation payments required to be made to an injured party, pursuant to certain provisions of the Israeli Securities Law. |
Under the Israeli Companies Law, a company may insure an office holder against the following liabilities incurred for acts performed by him or her as an office holder if and to the extent provided in the Company's articles of association:
| ● | a breach of the duty of loyalty to us, provided that the office holder acted in good faith and had a reasonable basis to believe that the act would not harm us; |
| ● | a breach of duty of care to us or to a third party; and |
| ● | a financial liability imposed on the office holder in favor of a third party. |
Subject to the provisions of the Companies Law and the Securities Law, we may also enter into a contract to insure an office holder, in respect of expenses, including reasonable litigation expenses and legal fees, incurred by an office holder in relation to an administrative proceeding instituted against such office holder or payment required to be made to an injured party, pursuant to certain provisions of the Securities Law.
Nevertheless, under the Israeli Companies Law, a company may not indemnify, exculpate or insure an office holder against any of the following:
| ● | a breach of fiduciary duty, except for indemnification and insurance for a breach of the duty of loyalty to us in the event office holder acted in good faith and had a reasonable basis to believe that the act would not prejudice us; |
| ● | a breach of duty of care committed intentionally or recklessly, excluding a breach arising out of the negligent conduct of the office holder; |
| ● | an act or omission committed with intent to derive unlawful personal benefit; or |
| ● | a fine, monetary sanction, penalty or forfeit levied against the office holder. |
Under the Israeli Companies Law, exculpation, indemnification and insurance of office holders require the approval of the compensation committee, board of directors and, in certain circumstances, the shareholders. Our amended and restated articles of association permit us to exculpate, indemnify and insure our office holders to the fullest extent permitted by the Israeli Companies Law.
| 93 |
Approval of Compensation to Our Officers
The Israeli Companies Law prescribes that compensation to officers must be approved by a company’s Board of Directors after obtaining the approval of the compensation committee.
As detailed above, our compensation committee consists of three independent directors: Israel Shamay, Gil Oren and Guy Regev. The responsibilities of the compensation committee are to set our overall policy on executive remuneration and to decide the specific remuneration, benefits and terms of employment for directors, officers and the Chief Executive Officer.
The objectives of the compensation committee’s policies are that such individuals should receive compensation which is appropriate given their performance, level of responsibility and experience. Compensation packages should also allow us to attract and retain executives of the necessary caliber while, at the same time, motivating them to achieve the highest level of corporate performance in line with the best interests of shareholders. In order to determine the elements and level of remuneration appropriate to each executive director, the compensation committee reviews surveys on executive pay, obtains external professional advice and considers individual performance.
Internal Auditor
Under the Israeli Companies Law, the board of directors must appoint an internal auditor, nominated by the audit committee. The role of the internal auditor is to examine, among other matters, whether our actions comply with the law and orderly business procedure. Under the Israeli Companies Law, an internal auditor may not be:
| ● | a person (or a relative of a person) who holds more than 5% of our shares; |
| ● | a person (or a relative of a person) who has the power to appoint a director or the general manager of the company; |
| ● | an executive officer or director of the company (or a relative thereof); or |
| ● | a member of our independent accounting firm, or anyone on his or her behalf. |
We comply with the requirement of the Israeli Companies Law relating to internal auditors. Our internal auditors examine whether our various activities comply with the law and orderly business procedure. Our current internal auditor is Deloitte.
D. Employees.
As of December 31, 2016, we had seven employees, three of whom were employed in management and administration, three of whom were employed in research and development and one of whom was employed in business development. All of these employees were located in Israel.
While none of our employees are party to any collective bargaining agreements, certain provisions of the collective bargaining agreements between the Histadrut (General Federation of Labor in Israel) and the Coordination Bureau of Economic Organizations (including the Industrialists’ Associations) are applicable to our employees by order of the Israel Ministry of Labor. These provisions primarily concern the length of the workday, minimum daily wages for professional workers, pension fund benefits for all employees, insurance for work-related accidents, procedures for dismissing employees, determination of severance pay and other conditions of employment. We generally provide our employees with benefits and working conditions beyond the required minimums. We have never experienced any employment-related work stoppages and believe our relationship with our employees is good.
| 94 |
E. Share Ownership.
The following table sets forth information regarding the beneficial ownership of our outstanding ordinary shares as of March 29, 2017 by the members of our senior management and board of directors individually and as a group. The beneficial ownership of ordinary shares is based on the 32,709,901 ordinary shares outstanding as of March 29, 2017 (which excludes 446,827 ordinary shares held in treasury) and is determined in accordance with the rules of the SEC and generally includes any ordinary shares over which a person exercises sole or shared voting or investment power. For purposes of the table below, we deem shares subject to options or warrants that are currently exercisable or exercisable within 60 days of March 29, 2017, to be outstanding and to be beneficially owned by the person holding the options or warrants for the purposes of computing the percentage ownership of that person but we do not treat them as outstanding for the purpose of computing the percentage ownership of any other person.
| Name of Beneficial Owner | Number
of | Percentage | ||||||
| Senior Management and Directors | ||||||||
| Ilan Cohn, PhD., Chairman of the Board | 214,852 | (1) | * | |||||
| Pnina Fishman, PhD., Chief Executive Officer and Director | 470,637 | (2) | 1.4 | % | ||||
| Motti Farbstein, Chief Operating Officer | 55,182 | (3) | * | |||||
| Guy Regev, Director | 54,240 | (4) | * | |||||
| Abraham Sartani, Ph.D, Director | 4,000 | (5) | * | |||||
| Gil Oren, Director | 9,166 | (6) | * | |||||
| Israel Shamay, Director | - | - | ||||||
| Senior Management and Directors as a group (7 persons) | 808,077 | 2.5 | % | |||||
| * | Denotes less than 1%
|
| (1) | Represents 133,567 ordinary shares, and (ii) 2,032,136 unregistered options to purchase 81,285 ordinary shares at an exercise price of NIS 1.247 per option and expire on May 9, 2017. All such warrants and options are fully vested. |
| (2) | Represents (i) 263,433 ordinary shares, (ii) 2,680,000 unregistered options to purchase 107,200 ordinary shares at an exercise price of NIS 0.644 per option and expiring on January 13, 2021 and (iii) 100,004 unregistered options to purchase 100,004 ordinary shares at an exercise price of NIS 3.573 per option and expire on October 22, 2025. Excludes 99,996 unregistered options to purchase 99,996 ordinary shares that vest in more than 60 days from March 29, 2017. |
| (3) | Represents (i) 1,257 ordinary shares, (ii) 754,387 unregistered options to purchase 30,175 ordinary shares, of which (1) 554,387 are exercisable into 22,175 ordinary shares at an exercise price of NIS 0.307 per option and expire on November 26, 2018, (2) 100,000 are exercisable into 4,000 ordinary shares at an exercise price of NIS 0.385 per option and expire on May 2, 2022, and (3) 100,000 are exercisable into 4,000 ordinary shares at an exercise price of NIS 0.326 per option and expire on March 20, 2023, and (iii) (1) 5,000 unregistered options to purchase 5,000 ordinary shares at an exercise price of NIS 8.1205 per option and expire on March 18, 2025, (2) 18,750 unregistered options to purchase 18,750 ordinary shares at an exercise price of NIS 4.317 per option and expire on February 18, 2026. Excludes 46,250 unregistered options to purchase 46,250 ordinary shares that vest in more than 60 days from March 29, 2017. |
| (4) | Represents (i) 24,240 ordinary shares, (ii) 250,000 registered warrants (Series 10) to purchase 10,000 ordinary shares at an exercise price of NIS 0.394 per warrant and expire on October 31, 2017, (iii) 250,000 registered warrants (Series 11) to purchase 10,000 ordinary shares at an exercise price of NIS 0.392 per warrant and expiring on April 31, 2017, and (iv) 250,000 unregistered options are exercisable into 10,000 ordinary shares at an exercise price of NIS 0.60 per option and expire on May 2, 2023. All such warrants and options are fully vested. |
| (5) | Represents 100,000 unregistered options to purchase 4,000 ordinary shares at an exercise price of NIS 0.385 per option and expire on August 14, 2022. All such options are fully vested. |
| (6) | Represents 9,166 unregistered options to purchase 9,166 ordinary shares at an exercise price of NIS 12 per option and expire on July 14, 2024. Excludes 834 unregistered options to purchase 834 ordinary shares that vest in more than 60 days from March 29, 2017. |
| 95 |
Share Option Plans
We maintain the following share option plans for our and our subsidiary’s employees, directors and consultants. In addition to the discussion below, see Note 12b of our consolidated financial statements, included in “Item 18. Financial Statements.”
Our Board of Directors administers our share option plans and has the authority to designate all terms of the options granted under our plans including the grantees, exercise prices, grant dates, vesting schedules and expiration dates, which may be no more than ten years after the grant date. Options may not be granted with an exercise price of less than the fair market value of our ordinary shares on the date of grant, unless otherwise determined by our Board of Directors.
As of December 31, 2016, options to purchase an aggregate of 737,028 ordinary shares, par value NIS 0.25, are outstanding pursuant to the 2003 and 2013 share option plans, or the 2003 and 2013 Plans.
2003 Share Option Plan
Under the 2003 Plan we granted options during the period between 2003 and 2013, at exercise prices between NIS 0.25 and NIS 31.175 per ordinary share, par value NIS 0.25. Options to purchase up to 1,132,514 ordinary shares, par value NIS 0.25, were available to be granted under the 2003 Plan. As of December 31, 2016, 7,675,728 options to purchase 307,028 ordinary shares were outstanding. Options granted to Israeli employees were in accordance with section 102 of the Income Tax Ordinance, 1961, or the Tax Ordinance, under the capital gains option set forth in section 102(b)(2) of the Tax Ordinance. The options are non-transferable.
The option term is for a period of ten years from the grant date. The options were granted for no consideration. The options vest over a four or two year period. As of December 31, 2016, options to purchase 307,028 ordinary shares, par value NIS 0.25, were fully vested.
2013 Share Option Plan
Under the 2013 Plan we granted options at exercise prices between NIS 3.573 and NIS 12 per ordinary share, par value NIS 0.25. Options to purchase up to 966,634 ordinary shares, par value NIS 0.25, were available to be granted under the 2013 Plan. As of December 31, 2016, 430,000 options to purchase 430,000 ordinary shares were outstanding. Options granted to Israeli employees were in accordance with section 102 of the Income Tax Ordinance, 1961, or the Tax Ordinance, under the capital gains option set forth in section 102(b)(2) of the Tax Ordinance. The options are non-transferable.
The option term is for a period of ten years from the grant date. The options were granted for no consideration. The options vest over a four year period. As of December 31, 2016, options to purchase 95,472 ordinary shares, par value NIS 0.25, were fully vested.
ITEM 7. Major Shareholders and Related Party Transactions
A. Major Shareholders.
To our knowledge, as of March 29, 2017 there were no shareholders who beneficially own more than 5% of our ordinary shares.
To our knowledge, as of March 29, 2017, we had 2 holders of record of our ADSs with a U.S. address, including Cede & Co., the nominee of The Depository Trust Company. These holders held in the aggregate 20,554,486 ordinary shares (represented by ADSs), or 62% of our outstanding ordinary shares as of March 29, 2017. The number of record holders in the United States is not representative of the number of beneficial holders of our ADSs or ordinary shares nor is it representative of where such beneficial holders are resident since many of these ADSs or ordinary shares were held by brokers or other nominees.
| 96 |
B. Related Party Transactions.
The following is a description of the transactions with related parties to which we, or our subsidiaries, are party, and which were in effect within the past three fiscal years. The descriptions provided below are summaries of the terms of such agreements, do not purport to be complete and are qualified in their entirety by the complete agreements.
We believe that we have executed all of our transactions with related parties on terms no less favorable to us than those we could have obtained from unaffiliated third parties. We are required by Israeli law to ensure that all future transactions between us and our officers, directors and principal shareholders and their affiliates are approved by a majority of our Board of Directors, including a majority of the independent and disinterested members of our Board of Directors, and that they are on terms no less favorable to us than those that we could obtain from unaffiliated third parties.
Employment and Consulting Agreements
We have or have had employment, consulting or related agreements with each member of our senior management. See “Item 6. Directors, Senior Management and Employees—Compensation”.
Indemnification Agreements
Our Articles of Association permit us to exculpate, indemnify and insure our directors and officeholders to the fullest extent permitted by the Israeli Companies Law. We have obtained directors’ and officers’ insurance for each of our officers and directors and have entered into indemnification agreements with all of our current officers and directors.
Agreements with Subsidiaries
See “Item 10. Additional Information—Material Contracts—OphthaliX Agreements” for a description of agreements with OphthaliX and Eye-Fite.
C. Interests of Experts and Counsel.
Not applicable.
A. Consolidated Financial Statements and Other Financial Information
See “Item 18. Financial Statements” for a list of all financial statements filed as part of this Annual Report on Form 20-F.
Legal Matters
Except as set forth below, we are not involved in any legal or arbitration proceedings that may have or have had in the recent past, significant effects on our financial position or profitability.
On June 29, 2015, we received a lawsuit, filed with the District Court of Tel-Aviv, requesting recognition of this lawsuit as a class action. The lawsuit named the Company, its Chief Executive Officer and directors as defendants. The lawsuit alleges, among other things, that we misled the public with regard to disclosures concerning the efficacy of our drug candidate, Piclidenoson. The claimant alleges that he suffered personal damages of over NIS 73,000, while also claiming that our shareholders suffered damages of approximately NIS 125 million. On March 31, 2016, we filed a response to the lawsuit. On March 1, 2017, a hearing was held in the District Court on whether to certify the lawsuit as a class action. A final hearing on the certification is scheduled for April 26, 2017. We strongly believe the lawsuit and its allegations to be baseless and without merit, and will vigorously defend this action.
Dividend Policy
We have never declared or paid cash dividends to our shareholders. Currently we do not intend to pay cash dividends. We intend to reinvest any earnings in developing and expanding our business. Any future determination relating to our dividend policy will be at the discretion of our Board of Directors and will depend on a number of factors, including future earnings, our financial condition, operating results, contractual restrictions, capital requirements, business prospects, applicable Israeli law and other factors our Board of Directors may deem relevant.
| 97 |
B. Significant Changes
See “Note 19:- Subsequent Events” to our consolidated financial statements included in this Annual Report beginning on page F-1 for a discussion of significant events that have occurred since December 31, 2016.
A. Offer and Listing Details
Ordinary Shares
Our ordinary shares have been trading on the Tel Aviv Stock Exchange, or TASE, under the symbol “CFBI” since October 2005.
The following table sets forth, for the periods indicated, the reported high and low closing sale prices of our ordinary shares on the TASE in NIS and U.S. dollars. U.S. dollar per ordinary share amounts were calculated using the U.S. dollar representative rate of exchange on the date to which the high or low market price is applicable, as reported by the Bank of Israel. As of December 31, 2016, we had 27,709,901 ordinary shares outstanding (which excludes 446,827 ordinary shares held in treasury). See “Item 10—Additional Information—Memorandum and Articles of Association” for a detailed description of the rights attaching to the shares.
We effected a 1-for-25 reverse share split with respect to our ordinary shares, options and warrants on May 12, 2013. Reported prices in the table below have been adjusted to give retroactive effect to the share split.
| NIS | U.S.$ | |||||||||||||||
Price Per Ordinary Share (1) | Price Per Ordinary Share (1) | |||||||||||||||
| High | Low | High | Low | |||||||||||||
| Annual: | ||||||||||||||||
| 2016 | 6.296 | 3.832 | 1.665 | 0.962 | ||||||||||||
| 2015 | 10.990 | 2.947 | 2.735 | 0.760 | ||||||||||||
| 2014 | 11.140 | 4.495 | 3.198 | 1.175 | ||||||||||||
| 2013 | 15.600 | 6.217 | 4.453 | 1.725 | ||||||||||||
| 2012 | 12.400 | 7.325 | 3.225 | 1.800 | ||||||||||||
| Quarterly: | ||||||||||||||||
| First Quarter 2017 (through March 29, 2017) | 4.688 | 3.448 | 1.230 | 0.946 | ||||||||||||
| Fourth Quarter 2016 | 4.949 | 3.959 | 1.310 | 1.041 | ||||||||||||
| Third Quarter 2016 | 5.132 | 4.052 | 1.335 | 1.059 | ||||||||||||
| Second Quarter 2016 | 6.296 | 4.928 | 1.665 | 1.291 | ||||||||||||
| First Quarter 2016 | 5.841 | 3.832 | 1.497 | 0.962 | ||||||||||||
| Fourth Quarter 2015 | 9.519 | 5.243 | 2.482 | 1.346 | ||||||||||||
| Third Quarter 2015 | 10.020 | 2.947 | 2.543 | 0.760 | ||||||||||||
| Second Quarter 2015 | 5.800 | 4.145 | 1.498 | 1.055 | ||||||||||||
| First Quarter 2015 | 10.990 | 4.554 | 2.735 | 1.144 | ||||||||||||
| Most Recent Six Months: | ||||||||||||||||
| March (through March 29, 2017) | 3.900 | 3.448 | 1.057 | 0.946 | ||||||||||||
| February | 4.000 | 3.566 | 1.067 | 0.946 | ||||||||||||
| January 2017 | 4.688 | 3.589 | 1.239 | 0.952 | ||||||||||||
| December 2016 | 4.563 | 3.959 | 1.187 | 1.041 | ||||||||||||
| November 2016 | 4.582 | 4.284 | 1.197 | 1.114 | ||||||||||||
| October 2016 | 4.949 | 4.548 | 1.310 | 1.179 | ||||||||||||
| September 2016 | 5.014 | 4.415 | 1.335 | 1.170 | ||||||||||||
| (1) | We effected a 1-for-25 reverse share split with respect to our ordinary shares, options and warrants on May 12, 2013. Reported prices in the table below have been adjusted to give retroactive effect to the share split. |
| 98 |
On March 29, 2017, the last reported sales price of our ordinary shares on the TASE was NIS 3.549 per share, or $0.979 per share. On March 29, 2017, the exchange rate of the NIS to the dollar was $1.00 = NIS 3.625 as reported by the Bank of Israel.
For information with respect to our warrants, see “Item 5. Operating and Financial Review and Prospects—Warrants”.
ADSs
On October 2, 2012, our ADSs began trading over the counter, or OTC, in the United States under the symbol “CANFY” and on November 19, 2013, our ADSs began trading on the NYSE MKT under the symbol “CANF.” One ADS represents two ordinary shares. See “Item 12—Description of Securities Other Than Equity Securities—American Depositary Shares” for a description of the rights attaching to the ADSs.
The following table sets forth, for the periods indicated, the reported high and low closing sale prices of our ADSs on the OTC and Nasdaq Capital Market in U.S. dollars.
| U.S.$ | ||||||||
| Price Per ADS (1) | ||||||||
| High | Low | |||||||
| Annual: | ||||||||
| 2016 | 3.35 | 1.95 | ||||||
| 2015 | 5.54 | 1.61 | ||||||
| 2014 | 6.50 | 2.41 | ||||||
| 2013 | 8.60 | 3.30 | ||||||
| 2012 (from October 2, 2012) | 5.50 | 4.74 | ||||||
| Quarterly: | ||||||||
| First Quarter 2017 (through March 29, 2017) | 2.45 | 1.79 | ||||||
| Fourth Quarter 2016 | 2.68 | 2.00 | ||||||
| Third Quarter 2016 | 2.72 | 2.07 | ||||||
| Second Quarter 2016 | 3.35 | 2.51 | ||||||
| First Quarter 2016 | 2.93 | 1.95 | ||||||
| Fourth Quarter 2015 | 4.66 | 2.64 | ||||||
| Third Quarter 2015 | 5.24 | 1.61 | ||||||
| Second Quarter 2015 | 3.29 | 1.95 | ||||||
| First Quarter 2015 | 5.54 | 2.20 | ||||||
| Most Recent Six Months: | ||||||||
| March 2017 (through March 29, 2017) | 2.06 | 1.87 | ||||||
| February 2017 | 2.02 | 1.83 | ||||||
| January 2017 | 2.45 | 1.79 | ||||||
| December 2016 | 2.46 | 2.00 | ||||||
| November 2016 | 2.38 | 2.13 | ||||||
| October 2016 | 2.68 | 2.33 | ||||||
| September 2016 | 2.72 | 2.28 | ||||||
| (1) | We effected a 1-for-25 reverse share split with respect to our ordinary shares, options and warrants on May 12, 2013. Reported prices in the table below have been adjusted to give retroactive effect to the share split. |
On March 29, 2017, the last reported sales price of our ADSs on the NYSE MKT was $1.91 per share.
| 99 |
B. Plan of Distribution.
Not applicable.
C. Markets.
See “—Offer and Listing Details” above.
D. Selling Shareholders.
Not applicable.
E. Dilution.
Not applicable.
F. Expenses of the Issue.
Not applicable.
ITEM 10. Additional Information
A. Share Capital.
Not applicable.
B. Memorandum and Articles of Association.
Our number with the Israeli Registrar of Companies is 512022153. Our purpose is set forth in Section 3 of our Articles of Association and includes every lawful purpose.
Our ordinary shares that are fully paid for are issued in registered form and may be freely transferred under our Articles of Association, unless the transfer is restricted or prohibited by applicable law or the rules of a stock exchange on which the shares are traded. The ownership or voting of our ordinary shares by non-residents of Israel is not restricted in any way by our Articles of Association or the laws of the State of Israel, except for ownership by nationals of some countries that are, or have been, in a state of war with Israel.
Pursuant to the Israeli Companies Law and our Articles of Association, our Board of Directors may exercise all powers and take all actions that are not required under law or under our Articles of Association to be exercised or taken by our shareholders, including the power to borrow money for company purposes.
Our Articles of Association enable us to increase or reduce our share capital. Any such changes are subject to the provisions of the Israeli Companies Law and must be approved by a resolution duly passed by our shareholders at a general or special meeting by voting on such change in the capital. In addition, transactions that have the effect of reducing capital, such as the declaration and payment of dividends in the absence of sufficient retained earnings and profits and an issuance of shares for less than their nominal value, require a resolution of our Board of Directors and court approval.
Dividends
We may declare a dividend to be paid to the holders of our ordinary shares in proportion to their respective shareholdings. Under the Israeli Companies Law, dividend distributions are determined by the board of directors and do not require the approval of the shareholders of a company unless such company’s articles of association provide otherwise. Our Articles of Association do not require shareholder approval of a dividend distribution and provide that dividend distributions may be determined by our Board of Directors.
Pursuant to the Israeli Companies Law, we may only distribute dividends from our profits accrued over the previous two years, as defined in the Israeli Companies Law, according to our then last reviewed or audited financial reports, or we may distribute dividends with court approval. In each case, we are only permitted to pay a dividend if there is no reasonable concern that payment of the dividend will prevent us from satisfying our existing and foreseeable obligations as they become due.
| 100 |
Election of Directors
Our ordinary shares do not have cumulative voting rights in the election of directors. As a result, the holders of a majority of the voting power represented at a shareholders meeting have the power to elect all of our directors, subject to the special approval requirements for external directors described under “Item 6. Directors, Senior Management and Employees — Board Practices — External Directors.”
Pursuant to our Articles of Association, other than the external directors, for whom special election requirements apply under the Israeli Companies Law, our directors are elected at a general or special meeting of our shareholders and serve on the Board of Directors until the end of the next general meeting or they are removed by the majority of our shareholders at a general or special meeting of our shareholders or upon the occurrence of certain events, in accordance with the Israeli Companies Law and our Articles of Association. In addition, our Articles of Association allow our Board of Directors to appoint directors to fill vacancies on the Board of Directors to serve until the next general meeting or special meeting, or earlier if required by our Articles of Association or applicable law. We have held elections for each of our non-external directors at each annual meeting of our shareholders since our initial public offering in Israel. External directors are elected for an initial term of three years and may be removed from office pursuant to the terms of the Israeli Companies Law. See “Item 6. Directors, Senior Management and Employees — Board Practices — External Directors.”
Shareholder Meetings
Under Israeli Companies Law, we are required to hold an annual general meeting of our shareholders once every calendar year that must be no later than 15 months after the date of the previous annual general meeting. All meetings other than the annual general meeting of shareholders are referred to as special meetings. Our Board of Directors may call special meetings whenever it sees fit, at such time and place, within or outside of Israel, as it may determine. In addition, the Israeli Companies Law and our Articles of Association provide that our Board of Directors is required to convene a special meeting upon the written request of (i) any two of our directors or one quarter of our Board of Directors or (ii) one or more shareholders holding, in the aggregate, either (1) 5% of our outstanding shares and 1% of our outstanding voting power or (2) 5% of our outstanding voting power.
Subject to the provisions of the Israeli Companies Law and the regulations promulgated thereunder, shareholders entitled to participate and vote at general meetings are the shareholders of record on a date to be decided by the board of directors, which may be between four and forty days prior to the date of the meeting. Furthermore, the Israeli Companies Law and our Articles of Association require that resolutions regarding the following matters must be passed at a general meeting of our shareholders:
| ● | amendments to our Articles of Association; |
| ● | appointment or termination of our auditors; |
| ● | appointment of directors and appointment and dismissal of external directors; |
| ● | approval of acts and transactions requiring general meeting approval pursuant to the Israeli Companies Law; |
| ● | director compensation, indemnification and change of the principal executive officer; |
| ● | increases or reductions of our authorized share capital; |
| ● | a merger; and |
| ● | the exercise of our Board of Director’s powers by a general meeting, if our Board of Directors is unable to exercise its powers and the exercise of any of its powers is required for our proper management. |
The Israeli Companies Law requires that a notice of any annual or special shareholders meeting be provided at least 21 days prior to the meeting and if the agenda of the meeting includes the appointment or removal of directors, the approval of transactions with office holders or interested or related parties, or an approval of a merger, notice must be provided at least 35 days prior to the meeting.
| 101 |
The Israeli Companies Law does not allow shareholders of publicly traded companies to approve corporate matters by written consent. Consequently, our Articles of Association does not allow shareholders to approve corporate matters by written consent.
Pursuant to our Articles of Association, holders of our ordinary shares have one vote for each ordinary share held on all matters submitted to a vote before the shareholders at a general meeting.
Quorum
The quorum required for our general meetings of shareholders consists of at least two shareholders present in person, by proxy or written ballot who hold or represent between them at least 25% of the total outstanding voting rights.
A meeting adjourned for lack of a quorum is adjourned to the same day in the following week at the same time and place or on a later date if so specified in the summons or notice of the meeting. At the reconvened meeting, any number of our shareholders present in person or by proxy shall constitute a lawful quorum.
Resolutions
Our Articles of Association provide that all resolutions of our shareholders require a simple majority vote, unless otherwise required by applicable law.
Israeli law provides that a shareholder of a public company may vote in a meeting and in a class meeting by means of a written ballot in which the shareholder indicates how he or she votes on resolutions relating to the following matters:
| ● | an appointment or removal of directors; |
| ● | an approval of transactions with office holders or interested or related parties; |
| ● | an approval of a merger or any other matter in respect of which there is a provision in the articles of association providing that decisions of the general meeting may also be passed by written ballot; |
| ● | authorizing the chairman of the board of directors or his relative to act as our chief executive officer or act with such authority; or authorize our chief executive officer or his relative to act as the chairman of the board of directors or act with such authority; and |
| ● | other matters which may be prescribed by Israel’s Minister of Justice. |
The provision allowing the vote by written ballot does not apply where the voting power of the controlling shareholder is sufficient to determine the vote. Our Articles of Association provides that our Board of Directors may prevent voting by means of a written ballot and this determination is required to be stated in the notice convening the general meeting.
The Israeli Companies Law provides that a shareholder, in exercising his or her rights and performing his or her obligations toward the company and its other shareholders, must act in good faith and in a customary manner, and avoid abusing his or her power. This is required when voting at general meetings on matters such as changes to the articles of association, increasing our registered capital, mergers and approval of related party transactions. A shareholder also has a general duty to refrain from depriving any other shareholder of its rights as a shareholder. In addition, any controlling shareholder, any shareholder who knows that its vote can determine the outcome of a shareholder vote and any shareholder who, under such company’s articles of association, can appoint or prevent the appointment of an office holder, is required to act with fairness towards the company. The Israeli Companies Law does not describe the substance of this duty except to state that the remedies generally available upon a breach of contract will also apply to a breach of the duty to act with fairness, and, to the best of our knowledge, there is no binding case law that addresses this subject directly.
Under the Israeli Companies Law, unless provided otherwise in a company’s articles of association, a resolution at a shareholders meeting requires approval by a simple majority of the voting rights represented at the meeting, in person, by proxy or written ballot, and voting on the resolution. A resolution for the voluntary winding up of the company requires the approval of holders of 75% of the voting rights represented at the meeting, in person, by proxy or by written ballot and voting on the resolution.
| 102 |
In the event of our liquidation, after satisfaction of liabilities to creditors, our assets will be distributed to the holders of our ordinary shares in proportion to their shareholdings. This right, as well as the right to receive dividends, may be affected by the grant of preferential dividend or distribution rights to the holders of a class of shares with preferential rights that may be authorized in the future.
Access to Corporate Records
Under
the Israeli Companies Law, all shareholders of a company generally have the right to review minutes of our general meetings, its
shareholders register and principal shareholders register, articles of association, financial statements and any document it is
required by law to file publicly with the Israeli Companies Registrar and the ISA. Any of our shareholders may request access
to review any document in our possession that relates to any action or transaction with a related party, interested party or office
holder that requires shareholder approval under the Israeli Companies Law. We may deny a request to review a document if we determine
that the request was not made in good faith, that the document contains a commercial secret or a patent or that the document’s
disclosure may otherwise prejudice our interests.
Acquisitions under Israeli Law
Full Tender Offer
A person wishing to acquire shares of a public Israeli company and who would as a result hold over 90% of the target company’s issued and outstanding share capital is required by the Israeli Companies Law to make a tender offer to all of our shareholders for the purchase of all of the issued and outstanding shares of the company. A person wishing to acquire shares of a public Israeli company and who would as a result hold over 90% of the issued and outstanding share capital of a certain class of shares is required to make a tender offer to all of the shareholders who hold shares of the same class for the purchase of all of the issued and outstanding shares of the same class. If the shareholders who do not accept the offer hold less than 5% of the issued and outstanding share capital of the company or of the applicable class, all of the shares that the acquirer offered to purchase will be transferred to the acquirer by operation of law (provided that a majority of the offerees that do not have a personal interest in such tender offer shall have approved the tender offer except that if the total votes to reject the tender offer represent less than 2% of the company’s issued and outstanding share capital, in the aggregate, approval by a majority of the offerees that do not have a personal interest in such tender offer is not required to complete the tender offer). However, a shareholder that had its shares so transferred may petition the court within six months from the date of acceptance of the full tender offer, whether or not such shareholder agreed to the tender or not, to determine whether the tender offer was for less than fair value and whether the fair value should be paid as determined by the court unless the acquirer stipulated in the tender offer that a shareholder that accepts the offer may not seek appraisal rights. If the shareholders who did not accept the tender offer hold 5% or more of the issued and outstanding share capital of the company or of the applicable class, the acquirer may not acquire shares of the company that will increase its holdings to more than 90% of our issued and outstanding share capital or of the applicable class from shareholders who accepted the tender offer.
Special Tender Offer
The Israeli Companies Law provides that an acquisition of shares of a public Israeli company must be made by means of a special tender offer if as a result of the acquisition the purchaser would become a holder of 25% or more of the voting rights in the company, unless one of the exemptions in the Israeli Companies Law is met. This rule does not apply if there is already another holder of at least 25% of the voting rights in the company. Similarly, the Israeli Companies Law provides that an acquisition of shares in a public company must be made by means of a tender offer if as a result of the acquisition the purchaser would become a holder of 45% or more of the voting rights in the company, if there is no other shareholder of the company who holds 45% or more of the voting rights in the company, unless one of the exemptions in the Israeli Companies Law is met.
A special tender offer must be extended to all shareholders of a company but the offeror is not required to purchase shares representing more than 5% of the voting power attached to our outstanding shares, regardless of how many shares are tendered by shareholders. A special tender offer may be consummated only if (i) at least 5% of the voting power attached to our outstanding shares will be acquired by the offeror and (ii) the number of shares tendered in the offer exceeds the number of shares whose holders objected to the offer.
| 103 |
If a special tender offer is accepted, then the purchaser or any person or entity controlling it or under common control with the purchaser or such controlling person or entity may not make a subsequent tender offer for the purchase of shares of the target company and may not enter into a merger with the target company for a period of one year from the date of the offer, unless the purchaser or such person or entity undertook to effect such an offer or merger in the initial special tender offer.
Merger
The Israeli Companies Law permits merger transactions if approved by each party’s board of directors and, unless certain requirements described under the Israeli Companies Law are met, a majority of each party’s shares voted on the proposed merger at a shareholders’ meeting called with at least 35 days’ prior notice.
For purposes of the shareholder vote, unless a court rules otherwise, the merger will not be deemed approved if a majority of the shares represented at the shareholders meeting that are held by parties other than the other party to the merger, or by any person who holds 25% or more of the outstanding shares or the right to appoint 25% or more of the directors of the other party, vote against the merger. If the transaction would have been approved but for the separate approval of each class or the exclusion of the votes of certain shareholders as provided above, a court may still approve the merger upon the request of holders of at least 25% of the voting rights of a company, if the court holds that the merger is fair and reasonable, taking into account the value of the parties to the merger and the consideration offered to the shareholders.
Upon the request of a creditor of either party to the proposed merger, the court may delay or prevent the merger if it concludes that there exists a reasonable concern that, as a result of the merger, the surviving company will be unable to satisfy the obligations of any of the parties to the merger, and may further give instructions to secure the rights of creditors.
In addition, a merger may not be completed unless at least 50 days have passed from the date that a proposal for approval of the merger was filed by each party with the Israeli Registrar of Companies and 30 days have passed from the date the merger was approved by the shareholders of each party.
Antitakeover Measures
The Israeli Companies Law allows us to create and issue shares having rights different from those attached to our ordinary shares, including shares providing certain preferred rights, distributions or other matters and shares having preemptive rights. As of the date of this annual report, we do not have any authorized or issued shares other than our ordinary shares. In the future, if we do create and issue a class of shares other than ordinary shares, such class of shares, depending on the specific rights that may be attached to them, may delay or prevent a takeover or otherwise prevent our shareholders from realizing a potential premium over the market value of their ordinary shares. The authorization of a new class of shares will require an amendment to our Articles of Association which requires the prior approval of the holders of a majority of our shares at a general meeting. In addition, the rules and regulations of the TASE also limit the terms permitted with respect to a new class of shares and prohibit any such new class of shares from having voting rights. Shareholders voting in such meeting will be subject to the restrictions provided in the Israeli Companies Law as described above.
Borrowing Powers
Under the Israeli Companies Law and our amended and restated articles of association, our Board of Directors may exercise all powers and take all actions that are not required under law or under our amended and restated articles of association to be exercised or taken by our shareholders or other corporate bodies, including the power to borrow money for company purposes.
Changes in Capital
Our amended and restated articles of association enable us to increase or reduce our share capital. Any such changes are subject to the provisions of the Israeli Companies Law and must be approved by a resolution duly passed by our shareholders at a general meeting by voting on such change in the capital. In addition, transactions that have the effect of reducing capital, such as the declaration and payment of dividends in the absence of sufficient retained earnings or profits and, in certain circumstances, an issuance of shares for less than their nominal value, require the approval of both our Board of Directors and an Israeli court.
| 104 |
C. Material Contracts.
The following are summary descriptions of certain material agreements to which we are a party. The descriptions provided below do not purport to be complete and are qualified in their entirety by the complete agreements, which are attached as exhibits to this Annual Report on Form 20-F.
OphthaliX Agreements
On November 21, 2011, we consummated a series of transactions resulting in the acquisition of 82.3% of the issued and outstanding share capital of OphthaliX, Inc., a Delaware corporation (formerly, Denali Concrete Management Inc., a Nevada corporation), whose common shares are traded in the United States on the OTC under the symbol “OPLI”.
The transactions were consummated pursuant to a series of agreements that we executed on November 21, 2011 with OphthaliX to spin-off our activity in the ophthalmology field to OphthaliX, or the Spin-Off Agreements. Prior to entering into the Spin-Off Agreements, we obtained a pre-ruling from the Israeli Tax Authority which prohibits us from selling more than 10% of the OphthaliX common stock that we hold until at least November 21, 2013. If we sell any of such shares prior to such date, we will be subject to a significant tax by the Israeli Tax Authority. As of December 31, 2016, we did not sell any of such shares.
Spin-Off Agreements
Pursuant to the Spin-Off Agreements, we formed Eye-Fite as a wholly-owned subsidiary of ours and transferred to all of the issued and outstanding share capital of Eye-Fite to OphthaliX, such that Eye-Fite became a wholly-owned subsidiary of OphthaliX. In consideration for the transfer of Eye-Fite, OphthaliX issued us 8,000,000 shares of OphthaliX common stock, which represented 86.7% of the issued and outstanding share capital of OphthaliX. In addition to the 8,000,000 shares of OphthaliX common stock that were issued to us in consideration for the transfer of Eye-Fite, we also acquired (i) 466,139 shares of OphthaliX common stock that were issued to us in exchange for 714,922 of our ordinary shares, which reflected a price of $5.148 per share of OphthaliX common stock, and (ii) 97,113 shares of OphthaliX common stock that were issued to us as consideration for our investment of $500,000 in OphthaliX, also at a price of $5.148 per share of OphthaliX common stock. We were also granted 1,267,316 warrants exercisable for 281,626 shares of OphthaliX common stock. Such warrants expired on November 20, 2016.
As a result of the Spin-Off Agreements, we appointed all of the members of the OphthaliX board of directors. According to the terms of the Spin-Off Agreements, OphthaliX will continue the development processes, clinical trials and registration of the ophthalmic indications of Piclidenoson.
As part of the acquisition transactions, OphthaliX raised approximately $3.33 million from a group of investors in a private placement of 646,776 shares of OphthaliX common stock, which represented approximately 6.2% of the issued and outstanding share capital of OphthaliX. As part of the private placement, Pnina Fishman, our Chief Executive Officer, invested $50,000 in OphthaliX and Guy Regev purchased shares of OphthaliX common stock from former OphthaliX shareholders for $75,000, each after approval by our audit committee and Board of Directors.
The acquisition transactions valued OphthaliX at approximately $50 million.
In connection with the acquisition transactions, we agreed not to withdraw any money from Eye-Fite or OphthaliX, except for the payments under the Services Agreement pursuant to which we are reimbursed for our costs plus 15%. See “—OphthaliX Agreements—Service Agreement”.
For additional information with respect to the Spin-Off Agreements, see “—OphthaliX Agreements—Service Agreement” and “Item 4. Information on the Company—Business Overview—Out-Licensing and Distribution Agreements—Eye-Fite Agreement”.
| 105 |
Services Agreement
On November 21, 2011, we entered into a services agreement, or the Services Agreement, with OphthaliX and Eye-Fite, pursuant to which we provide management services to OphthaliX and Eye-Fite with respect to (i) all pre-clinical and clinical research studies of Piclidenoson in the ophthalmic field, (ii) drug manufacturing and supply with respect to the compounds related to the Eye-Fite Agreement, (iii) QT studies in human beings, and (iv) payments to consultants that are listed in the Services Agreement for their involvement in the clinical trials and in all other activities necessary to launch Piclidenoson for the treatment of ophthalmic diseases. As consideration for the foregoing services, we will be reimbursed by OphthaliX for our costs and expenses incurred in rendering such services plus 15% (not including VAT, if applicable) and in relation to expenses and costs of intellectual property maintenance, we will “pass through” any such payments and expenses made to third parties and will receive reimbursement for such costs and expenses from OphthaliX. In addition, OphthaliX must abide by all current ongoing clinical trial agreements that we are party to and OphthaliX must pay all payments under those agreements from November 21, 2011 onwards. Further, we are entitled to an additional payment of 2.5%, or the additional payment, of any revenues received by OphthaliX and Eye-Fite in connection with the use of Piclidenoson in the ophthalmic field.
During the five-year period following the date of the execution of the Services Agreement, which ended on November 21, 2016, we were entitled to convert our right to the additional payment into a warrant to purchase 480,022 shares of OphthaliX common stock exercisable at $5.148 per share, representing approximately 5% of the shares of OphthaliX common stock on a fully diluted basis as of the date of closing of the Spin-Off Agreements and the Services Agreement. The Services Agreement is for an unlimited duration. However, following the first anniversary of the execution of the Services Agreement, each party is entitled to terminate the agreement if at least six months’ prior notice, or less with respect to termination for “cause”, as defined in the Services Agreement, is provided to the counterparty.
In February 2013, as last updated in August 2015, we sent a formal letter to OphthaliX, stating that we agree to defer the payments under the Services Agreement from January 31, 2013 for the performance of the clinical trials of Piclidenoson in ophthalmic indications until the completion of a fundraising by OphthaliX that will allow such payments. Also, in August 2015, we issued another financial support letter pursuant to which we committed to cover any shortfall in the costs and expenses of operations of OphthaliX which are in excess of OphthaliX's available cash to finance its operations, including cash generated from any future sale of Can-Fite shares . Any related balance on amounts owed bear interest at a rate of 3% per annum.
Both letters expired on October 10, 2016. On November 14, 2016, we agreed to extend the support letter under the same terms and conditions in order to fund our operations. Such letter expired on February 28, 2017. Deferred payments under the Services Agreement is currently due. As of December 31, 2016, the deferred payments to us totaled $4,459,000.
License Agreement
See “Item 4. Information on the Company—Out-Licensing and Distribution Agreements”.
Employment and Consulting Agreements
See “Item 6. Directors, Senior Management and Employees—Compensation—Employment and Consulting Agreements”.
D. Exchange Controls
There are no Israeli government laws, decrees or regulations that restrict or that affect our export or import of capital or the remittance of dividends, interest or other payments to non-resident holders of our securities, including the availability of cash and cash equivalents for use by us and our wholly-owned subsidiaries, except or otherwise as set forth under “Item 10.E. Additional Information — Taxation.”
E. Taxation
E. Taxation
Certain Israeli Tax Considerations
The following is a summary of the material Israeli tax laws applicable to us. This section also contains a discussion of material Israeli tax consequences concerning the ownership and disposition of our ordinary shares. This summary does not discuss all the aspects of Israeli tax law that may be relevant to a particular investor in light of his or her personal investment circumstances or to some types of investors subject to special treatment under Israeli law. Examples of this kind of investor include residents of Israel or traders in securities who are subject to special tax regimes not covered in this discussion. Because certain parts of this discussion are based on new tax legislation that has not yet been subject to judicial or administrative interpretation, we cannot assure you that the appropriate tax authorities or the courts will accept the views expressed in this discussion. The discussion does not cover all possible tax consequences.
| 106 |
You are urged to consult your own tax advisor as to the Israeli and other tax consequences of the purchase, ownership and disposition of our ADSs, including, in particular, the effect of any non-Israeli, state or local taxes.
General Corporate Tax Structure in Israel
Israeli companies are generally subject to corporate tax , which has decreased in recent years, from a rate of 26.5% in 2014 and 2015 to 25% in 2016 and to 24% in 2017, and will further decrease to 23% for 2018. However, the effective corporate tax rate payable by a company that derives income from an Approved Enterprise, a Privileged Enterprise or a Preferred Enterprise (as discussed below) may be considerably less. Capital gains generated by an Israeli company are generally subject to tax at the corporate tax rate.
In 2006, transfer pricing regulations came into force, following the introduction of Section 85A of the Israeli Tax Ordinance under Amendment 132. The transfer pricing rules require that cross-border transactions between related parties be carried out implementing an arms’ length principle and reported and taxed accordingly.
In 2008, the Knesset passed an amendment to the Income Tax (Inflationary Adjustments) Law, 1985, which limits the scope of the law starting in 2008 and thereafter. Starting in 2008, the revenues for tax purposes are measured in nominal values, excluding certain adjustments for changes in the consumer price index carried out in the period up to December 31, 2007. The amended law includes, among other provisions, the elimination of the inflationary additions and deductions and the additional deduction for depreciation for the period starting in 2008.
Pre-Ruling from the Israeli Income Tax Authorities
In connection with the Spin-Off, we received a pre-ruling decision from the Israeli Income Tax Authority which confirms: (i) that the grant of the license to Eye-Fite is not liable for tax pursuant to the provisions of section 104a to the Income Tax Ordinance (New Version), 1961, or the Ordinance; (ii) that OphthaliX is considered the receiving company pursuant to section 103c(7)(b) to the Ordinance; (iii) that the sale of Eye-Fite shares to OphthaliX as consideration for OphthaliX shares does not create liability for tax pursuant to the provisions of section 103t to the Ordinance, or change in structure; and (iv) the date for the change in structure was determined. According to the tax pre-ruling, the date of change in structure shall also be the date of exchange of shares with respect to the spin-off and notification to the tax assessor. We and Eye-Fite presented to the tax assessor and the merger and spin-off department of the tax assessor the forms required by the Ordinance and the regulations thereunder. The tax pre-ruling further provides that the grant of a license to Eye-Fite as consideration for the issuance of Eye-Fite shares to us does not create liability for tax pursuant to the provisions of section 104a to the Ordinance.
According to the pre-ruling, we must not sell more than 10% of our common stock holdings in OphthaliX issued in connection with the change in structure for at least two years from the date of the change (i.e., November 21, 2011), OphthaliX must not sell more than 10% of its ordinary share holdings in Eye-Fite received in connection with the change in structure for at least two years from the date of the change and Eye-Fite must retain the assets received from us in connection with the change in structure for at least two years from the date of the change.
The shares of Eye-Fite which were transferred to OphthaliX in connection with the change in structure will be held in escrow. The sale of these shares will be deemed as a sale by an Israeli company and will be taxed accordingly. The trustee will withhold tax at the source.
The shares of OphthaliX which were transferred to us in connection with the change in structure will be held in escrow. The sale of these shares will be deemed as a sale by an Israeli company and will be taxed accordingly. The trustee will withhold tax at the source.
Any dividend distributed by Eye-Fite to OphthaliX will be taxed in Israel in accordance with paragraph 125(b)5 of the Israeli Tax Ordinance.
A description of the terms of the pre-ruling is also included in the notes to the financial statements.
| 107 |
Tax Benefits and Grants for Research and Development
Israeli tax law allows, under certain conditions, a tax deduction for research and development expenditures, including capital expenditures, for the year in which they are incurred. These expenses must relate to scientific research and development projects and must be approved by the Office of the Chief Scientist, or the OCS, of the relevant Israeli government ministry, determined by the field of research. Furthermore, the research and development must be for the promotion of the company and carried out by or on behalf of the company seeking such tax deduction. The amount of such deductible expenses is reduced by the sum of any funds received through government grants for the funding of the scientific research and development projects. No deduction under these research and development deduction rules is allowed if such deduction is related to an expense invested in an asset depreciable under the general depreciation rules of the Tax Ordinance. Expenditures not so approved are deductible in equal amounts over three years.
On a yearly basis, we evaluate the applicability of the above tax deduction for research and development expenditures and, based on our evaluation, determine whether to apply to the OCS for approval of a tax deduction. There can be no assurance that any application for a tax deduction will be accepted.
Taxation of our Shareholders
Capital Gains Taxes Applicable to Non-Israeli Resident Shareholders. Shareholders that are not Israeli residents are generally exempt from Israeli capital gains tax on any gains derived from the sale, exchange or disposition of our shares, provided that such shareholders did not acquire their shares prior to our initial public offering on the TASE and such gains were not derived from a permanent establishment or business activity of such shareholders in Israel. However, non-Israeli corporations will not be entitled to the foregoing exemptions if an Israeli resident (i) has a controlling interest of 25% or more in such non-Israeli corporation or (ii) is the beneficiary of or is entitled to 25% or more of the revenues or profits of such non-Israeli corporation, whether directly or indirectly.
In addition, under the U.S.-Israel Income Tax Treaty, 1995, or the U.S.-Israel Tax Treaty, the sale, exchange or disposition of our shares by a shareholder who is a U.S. resident (for purposes of the U.S.-Israel Tax Treaty) holding the shares as a capital asset is exempt from Israeli capital gains tax unless either (i) the shareholder holds, directly or indirectly, shares representing 10% or more of our voting capital during any part of the 12-month period preceding such sale, exchange or disposition or (ii) the capital gains arising from such sale are attributable to a permanent establishment of the shareholder located in Israel. In either case, the sale, exchange or disposition of the shares would be subject to Israeli tax, to the extent applicable; however, under the U.S.-Israel Tax Treaty, the U.S. resident would be permitted to claim a credit for the tax against the U.S. federal income tax imposed with respect to the sale, exchange or disposition, subject to the limitations in U.S. laws applicable to foreign tax credits. The U.S.-Israel Tax Treaty does not relate to U.S. state or local taxes.
Shareholders may be required to demonstrate that they are exempt from tax on their capital gains in order to avoid withholding at source at the time of sale.
Taxation of Non-Israeli Shareholders on Receipt of Dividends. Non-residents of Israel are generally subject to Israeli income tax on the receipt of dividends paid on our shares at the rate of 20%, which tax will be withheld at the source, unless a different rate is provided in a tax treaty between Israel and the shareholder’s country of residence. With respect to a person who is a “substantial shareholder” at the time receiving the dividend or on any date in the 12 months preceding such date, the applicable tax rate is 25%. A “substantial shareholder” is generally a person who alone, or together with his relative or another person who collaborates with him on a permanent basis, holds, directly or indirectly, at least 10% of any of the “means of control” of the corporation. “Means of control” generally include the right to vote, receive profits, nominate a director or an officer, receive assets upon liquidation, or order someone who holds any of the aforesaid rights how to act, and all regardless of the source of such right. Under the U.S.-Israel Tax Treaty, the maximum rate of tax withheld in Israel on dividends paid to a holder of our ordinary shares who is a U.S. resident (for purposes of the U.S.-Israel Tax Treaty) is 25%. However, generally, the maximum rate of withholding tax on dividends that are paid to a U.S. corporation holding 10% or more of our outstanding voting capital throughout the tax year in which the dividend is distributed as well as the previous tax year is 12.5%.
A non-resident of Israel who receives dividends from which tax was withheld is generally exempt from the duty to file returns in Israel in respect of such income, provided such income was not derived from a business conducted in Israel by the taxpayer, and the taxpayer has no other taxable sources of income in Israel.
Taxation of Israeli Shareholders on Receipt of Dividends
Residents of Israel are generally subject to Israeli income tax on the receipt of dividends paid on our shares at the rate of 25%, which tax will be withheld at the source. With respect to a person who is a “substantial shareholder” at the time of receiving the dividend or on any date within the 12 months preceding such date, the applicable tax rate is 30%.
| 108 |
U.S. Federal Income Tax Consequences
The following is a general summary of what we believe to be material U.S. federal income tax consequences relating to the purchase, ownership and disposition of our ordinary shares and ADSs by U.S. Investors (as defined below) that hold such shares or ADSs as capital assets. This summary is based on the Internal Revenue Code, or the Code, the regulations of the U.S. Department of the Treasury issued pursuant to the Code, or the Treasury Regulations, and administrative and judicial interpretations thereof, all as in effect on the date hereof and all of which are subject to change, possibly with retroactive effect, or to different interpretation. No ruling has been sought from the IRS with respect to any United States federal income tax consequences described below, and there can be no assurance that the IRS or a court will not take a contrary position. This summary does not address all of the tax considerations that may be relevant to specific U.S. Investors in light of their particular circumstances or to U.S. Investors subject to special treatment under U.S. federal income tax law (such as banks, insurance companies, tax-exempt entities, retirement plans, regulated investment companies, partnerships, dealers in securities, brokers, real estate investment trusts, certain former citizens or residents of the United States, persons who acquire our shares or ADSs as part of a straddle, hedge, conversion transaction or other integrated investment, persons that have a “functional currency” other than the U.S. dollar, persons that own (or are deemed to own, indirectly or by attribution) 10% or more of our shares or ADSs or persons that generally mark their securities to market for U.S. federal income tax purposes). This summary does not address any U.S. state or local or non-U.S. tax considerations or any U.S. federal estate, gift or alternative minimum tax considerations or any U.S. federal tax consequences other than U.S. federal income tax consequences.
As used in this summary, the term “U.S. Investor” means a beneficial owner of our shares or ADSs that is, for U.S. federal income tax purposes, (i) an individual citizen or resident of the United States, (ii) a corporation, or other entity taxable as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the United States, any state thereof, or the District of Columbia, (iii) an estate the income of which is subject to U.S. federal income tax regardless of its source or (iv) a trust with respect to which a court within the United States is able to exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of its substantial decisions, or that has a valid election in effect under applicable Treasury Regulations to be treated as a “United States person.”
If an entity treated as a partnership for U.S. federal income tax purposes holds our shares or ADSs, the tax treatment of such partnership and each partner thereof will generally depend upon the status and activities of the partnership and such partner. A holder that is treated as a partnership for U.S. federal income tax purposes should consult its own tax advisor regarding the U.S. federal income tax considerations applicable to it and its partners of the purchase, ownership and disposition of its shares or ADSs.
Prospective investors should be aware that this summary does not address the tax consequences to investors who are not U.S. Investors. Prospective investors should consult their own tax advisors as to the particular tax considerations applicable to them relating to the purchase, ownership and disposition of their shares or ADSs, including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
Taxation of U.S. Investors
The discussions under “— Distributions” and under “— Sale, Exchange or Other Disposition of Ordinary Shares and ADSs” below assumes that we will not be treated as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes. We believe that we may be a PFIC during 2016 although we have not determined whether we will be a PFIC in 2017, or in any subsequent year, our operating results for any such years may cause us to be a PFIC. For a discussion of the rules that would apply if we are treated as a PFIC, see the discussion under “— Passive Foreign Investment Company.”
Distributions. We have no current plans to pay dividends. To the extent we pay any dividends, a U.S. Investor will be required to include in gross income as a taxable dividend the amount of any distributions made on the ordinary shares or ADSs , including the amount of any Israeli taxes withheld, to the extent that those distributions are paid out of our current or accumulated earnings and profits as determined for U.S. federal income tax purposes. Any distributions in excess of our earnings and profits will be applied against and will reduce the U.S. Investor's tax basis in its shares or ADSs and to the extent they exceed that tax basis, will be treated as gain from the sale or exchange of those shares or ADSs . If we were to pay dividends, we expect to pay such dividends in NIS with respect to the shares and in U.S. dollars with respect to ADSs. A dividend paid in NIS, including the amount of any Israeli taxes withheld, will be includible in a U.S. Investor's income as a U.S. dollar amount calculated by reference to the exchange rate in effect on the date such dividend is received, regardless of whether the payment is in fact converted into U.S. dollars. If the dividend is converted to U.S. dollars on the date of receipt, a U.S. Investor generally will not recognize a foreign currency gain or loss. However, if the U.S. Investor converts the NIS into U.S. dollars on a later date, the U.S. Investor must include, in computing its income, any gain or loss resulting from any exchange rate fluctuations. The gain or loss will be equal to the difference between (i) the U.S. dollar value of the amount included in income when the dividend was received and (ii) the amount received on the conversion of the NIS into U.S. dollars. Such gain or loss will generally be ordinary income or loss and United States source for U.S. foreign tax credit purposes. U.S. Investors should consult their own tax advisors regarding the tax consequences to them if we pay dividends in NIS or any other non-U.S. currency.
| 109 |
Subject to certain significant conditions and limitations, including potential limitations under the U.S.-Israel Tax Treaty, any Israeli taxes paid on or withheld from distributions from us and not refundable to a U.S. Investor may be credited against the investor’s U.S. federal income tax liability or, alternatively, may be deducted from the investor’s taxable income. This election is made on a year-by-year basis and applies to all foreign taxes paid by a U.S. Investor or withheld from a U.S. Investor that year. Dividends paid on the shares generally will constitute income from sources outside the United States and be categorized as “passive category income” or, in the case of some U.S. Investors, as “general category income” for U.S. foreign tax credit purposes.
Because the rules governing foreign tax credits are complex, U.S. Investors should consult their own tax advisor regarding the availability of foreign tax credits in their particular circumstances. In addition, the U.S. Treasury Department has expressed concerns that parties to whom ADSs are pre-released may be taking actions that are inconsistent with the claiming of foreign tax credits by U.S. holders of ADSs. Accordingly, the creditability of Israeli taxes could be affected by future actions that may be taken by the U.S. Treasury Department or parties to whom ADSs are pre-released.
Dividends paid on the shares and ADSs will not be eligible for the “dividends-received” deduction generally allowed to corporate U.S. Investors with respect to dividends received from U.S. corporations.
For taxable years beginning after December 31, 2012, certain distributions treated as dividends that are received by an individual U.S. Holder from “qualified foreign corporations” generally qualify for a 20% reduced maximum tax rate so long as certain holding period and other requirements are met. A non-US. corporation (other than a corporation that is treated as a PFIC for the taxable year in which the dividend is paid or the preceding taxable year) generally will be considered to be a qualified foreign corporation (i) if it is eligible for the benefits of a comprehensive tax treaty with the United States which the Secretary of Treasury of the United States determines is satisfactory for purposes of this provision and which includes an exchange of information program, or (ii) with respect to any dividend it pays on stock (or ADSs in respect of such stock) which is readily tradable on an established securities market in the United States. Dividends paid by us in a taxable year in which we are not a PFIC and with respect to which we were not a PFIC in the preceding taxable year are expected to be eligible for the 20% reduced maximum tax rate, although we can offer no assurances in this regard. However, any dividend paid by us in a taxable year in which we are a PFIC or were a PFIC in the preceding taxable year will be subject to tax at regular ordinary income rates. As mentioned above, we have not determined whether we are currently a PFIC or not.
Sale, Exchange or Other Disposition of Ordinary Shares and ADSs. Subject to the discussion under “— Passive Foreign Investment Company” below, a U.S. Investor generally will recognize capital gain or loss upon the sale, exchange or other disposition of our shares or ADSs in an amount equal to the difference between the amount realized on the sale, exchange or other disposition and the U.S. Investor's adjusted tax basis in such shares. This capital gain or loss will be long-term capital gain or loss if the U.S. Investor's holding period in our shares exceeds one year. Preferential tax rates for long-term capital gain (currently, with a maximum rate of % for taxable years beginning after December 31, 2012) will apply to individual U.S. Investors. The deductibility of capital losses is subject to limitations. The gain or loss will generally be income or loss from sources within the United States for U.S. foreign tax credit purposes, subject to certain exceptions in U.S.-Israel Tax Treaty.
U.S. Investors should consult their own tax advisors regarding the U.S. federal income tax consequences of receiving currency other than U.S. dollars upon the disposition of their shares or ADSs .
Passive Foreign Investment Company
In general, a corporation organized outside the United States will be treated as a PFIC for U.S. federal income tax purposes in any taxable year in which either (i) at least 75% of its gross income is “passive income” or (ii) on average at least 50% of its assets by value produce passive income or are held for the production of passive income. Passive income for this purpose generally includes, among other things, certain dividends, interest, royalties, rents and gains from commodities and securities transactions and from the sale or exchange of property that gives rise to passive income. Passive income also includes amounts derived by reason of the temporary investment of funds, including those raised in the public offering. In determining whether a non-U.S. corporation is a PFIC, a proportionate share of the income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account.
| 110 |
Under the tests described above, whether or not we are a PFIC will be determined annually based upon the composition of our income and the composition and valuation of our assets, all of which are subject to change.
We believe that we may be a PFIC during 2016 although we have not determined whether we will be a PFIC in 2017, or in any subsequent year, our operating results for any such years may cause us to be a PFIC.
U.S. Investors should be aware of certain tax consequences of investing directly or indirectly in us if we are a PFIC. A U.S. Investor is subject to different rules depending on whether the U.S. Investor makes an election to treat us as a “qualified electing fund,” known as a QEF election, for the first taxable year that the U.S. Investor holds shares or ADSs , which is referred to in this disclosure as a “timely QEF election,” makes a “mark-to-market” election with respect to the shares or ADSs (if such election is available), or makes neither election.
QEF Election. A U.S. Investor who makes a timely QEF election, referred to in this disclosure as an “Electing U.S. Investor,” with respect to us must report for U.S. federal income tax purposes his pro rata share of our ordinary earnings and net capital gain, if any, for our taxable year that ends with or within the taxable year of the Electing U.S. Investor. The “net capital gain” of a PFIC is the excess, if any, of the PFIC’s net long-term capital gains over its net short-term capital losses. The amount so included in income generally will be treated as ordinary income to the extent of such Electing U.S. Investor's allocable share of the PFIC’s ordinary earnings and as long-term capital gain to the extent of such Electing U.S. Investor's allocable share of the PFIC’s net capital gains. Such Electing U.S. Investor generally will be required to translate such income into U.S. dollars based on the average exchange rate for the PFIC’s taxable year with respect to the PFIC’s functional currency. Such income generally will be treated as income from sources outside the United States for U.S. foreign tax credit purposes. Amounts previously included in income by such Electing U.S. Investor under the QEF rules generally will not be subject to tax when they are distributed to such Electing U.S. Investor. The Electing U.S. Investor's tax basis in our shares or ADSs generally will increase by any amounts so included under the QEF rules and decrease by any amounts not included in income when distributed.
An Electing U.S. Investor will be subject to U.S. federal income tax on such amounts for each taxable year in which we are a PFIC, regardless of whether such amounts are actually distributed to such Electing U.S. Investor. However, an Electing U.S. Investor may, subject to certain limitations, elect to defer payment of current U.S. federal income tax on such amounts, subject to an interest charge. If an Electing U.S. Investor is an individual, any such interest will be treated as non-deductible “personal interest.”
Any net operating losses or net capital losses of a PFIC will not pass through to the Electing U.S. Investor and will not offset any ordinary earnings or net capital gain of a PFIC recognized by Electing U.S. Investor in subsequent years.
So long as an Electing U.S. Investor's QEF election with respect to us is in effect with respect to the entire holding period for our shares or ADSs , any gain or loss recognized by such Electing U.S. Investor on the sale, exchange or other disposition of such shares or ADSs generally will be long-term capital gain or loss if such Electing U.S. Investor has held such shares or ADSs for more than one year at the time of such sale, exchange or other disposition. Preferential tax rates for long-term capital gain (currently, a maximum rate of % for taxable years beginning after December 31, 2012) will apply to individual U.S. Investors. The deductibility of capital losses is subject to limitations.
In general, a U.S. Investor must make a QEF election on or before the due date for filing its income tax return for the first year to which the QEF election is to apply. A U.S. Investor makes a QEF election by completing the relevant portions of and filing IRS Form 8621 in accordance with the instructions thereto. Upon request, we will annually furnish U.S. Investors with information needed in order to complete IRS Form 8621 (which form would be required to be filed with the IRS on an annual basis by the U.S. Investor) and to make and maintain a valid QEF election for any year in which we or any of our subsidiaries that we control is a PFIC. There is no assurance, however, that we will have timely knowledge of our status as a PFIC, or that the information that we provide will be adequate to allow U.S. Investors to make a QEF election. A QEF election will not apply to any taxable year during which we are not a PFIC, but will remain in effect with respect to any subsequent taxable year in which we become a PFIC. Each U.S. Investor should consult its own tax advisor with respect to the advisability of, the tax consequences of, and the procedures for making a QEF election with respect to us.
| 111 |
Mark-to-Market Election. Alternatively, if our shares or ADSs are treated as “marketable stock,” a U.S. Investor would be allowed to make a “mark-to-market” election with respect to our shares or ADSs, provided the U.S. Investor completes and files IRS Form 8621 in accordance with the relevant instructions and related Treasury Regulations. If that election is made, the U.S. Investor generally would include as ordinary income in each taxable year the excess, if any, of the fair market value of our shares or ADSs at the end of the taxable year over such holder’s adjusted tax basis in such shares or ADSs. The U.S. Investor would also be permitted an ordinary loss in respect of the excess, if any, of the U.S. Investor's adjusted tax basis in our shares or ADSs over their fair market value at the end of the taxable year, but only to the extent of the net amount previously included in income as a result of the mark-to-market election. A U.S. Investor's tax basis in our shares or ADSs would be adjusted to reflect any such income or loss amount. Gain realized on the sale, exchange or other disposition of our shares or ADSs would be treated as ordinary income, and any loss realized on the sale, exchange or other disposition of our ordinary shares or ADSs would be treated as ordinary loss to the extent that such loss does not exceed the net mark-to-market gains previously included in income by the U.S. Investor, and any loss in excess of such amount will be treated as capital loss. Amounts treated as ordinary income will not be eligible for the favorable tax rates applicable to qualified dividend income or long-term capital gains.
Generally, stock will be considered marketable stock if it is “regularly traded” on a “qualified exchange” within the meaning of applicable Treasury Regulations. A class of stock is regularly traded on an exchange during any calendar year during which such class of stock is traded, other than in de minimis quantities, on at least 15 days during each calendar quarter. To be marketable stock, our shares and ADSs must be regularly traded on a qualifying exchange (i) in the United States that is registered with the SEC or a national market system established pursuant to the Exchange Act or (ii) outside the United States that is properly regulated and meets certain trading, listing, financial disclosure and other requirements. Our shares should constitute “marketable stock” as long as they remain listed on the OTC and/or the NYSE MKT and are regularly traded. Our ADSs will be listed on the OTC and/or the NYSE MKT. While we believe that our ADSs may be treated as marketable stock for purposes of the PFIC rules so long as they are listed on the OTC and/or the NYSE MKT and are regularly traded, the IRS has not provided a list of the exchanges that meet the foregoing requirements and thus no assurance can be provided that our ADSs will be (or will remain) treated as marketable stock for purposes of the PFIC rules.
A mark-to-market election will not apply to our shares or ADSs held by a U.S. Investor for any taxable year during which we are not a PFIC, but will remain in effect with respect to any subsequent taxable year in which we become a PFIC. Such election will not apply to any PFIC subsidiary that we own. Each U.S. Investor is encouraged to consult its own tax advisor with respect to the availability and tax consequences of a mark-to-market election with respect to our shares and ADSs.
Default PFIC Rules. A U.S. Investor who does not make a timely QEF election or a mark-to-market election, referred to in this disclosure as a “Non-Electing U.S. Investor,” will be subject to special rules with respect to (i) any “excess distribution” (generally, the portion of any distributions received by the Non-Electing U.S. Investor on the shares or ADSs in a taxable year in excess of 125% of the average annual distributions received by the Non-Electing U.S. Investor in the three preceding taxable years, or, if shorter, the Non-Electing U.S. Investor's holding period for the shares or ADSs ), and (ii) any gain realized on the sale or other disposition of such shares or ADSs . Under these rules:
| ● | the excess distribution or gain would be allocated ratably over the Non-Electing U.S. Investor's holding period for such shares or ADSs ; |
| ● | the amount allocated to the current taxable year and any year prior to us becoming a PFIC would be taxed as ordinary income; and |
| ● | the amount allocated to each of the other taxable years would be subject to tax at the highest rate of tax in effect for the applicable class of taxpayer for that year, and an interest charge for the deemed deferral benefit would be imposed with respect to the resulting tax attributable to each such other taxable year. |
If a Non-Electing U.S. Investor who is an individual dies while owning our shares or ADSs , the Non-Electing U.S. Investor's successor would be ineligible to receive a step-up in tax basis of such shares or ADSs . Non-Electing U.S. Investors should consult their tax advisors regarding the application of the PFIC rules to their specific situation.
A Non-Electing U.S. Investor who wishes to make a QEF election for a subsequent year may be able to make a special “purging election” pursuant to Section 1291(d) of the Code. Pursuant to this election, a Non-Electing U.S. Investor would be treated as selling his or her shares or ADSs for fair market value on the first day of the taxable year for which the QEF election is made. Any gain on such deemed sale would be subject to tax under the rules for Non-Electing U.S. Investors as discussed above. Non-Electing U.S. Investors should consult their tax advisors regarding the availability of a “purging election” as well as other available elections.
To the extent a distribution on our shares or ADSs does not constitute an excess distribution to a Non-Electing U.S. Investor, such Non-Electing U.S. Investor generally will be required to include the amount of such distribution in gross income as a dividend to the extent of our current or accumulated earnings and profits (as determined for U.S. federal income tax purposes) that are not allocated to excess distributions. The tax consequences of such distributions are discussed above under “— Taxation of U.S. Investors — Distributions.” Each U.S. Holder is encouraged to consult its own tax advisor with respect to the appropriate U.S. federal income tax treatment of any distribution on our shares.
| 112 |
If we are treated as a PFIC for any taxable year during the holding period of a Non-Electing U.S. Investor, we will continue to be treated as a PFIC for all succeeding years during which the Non-Electing U.S. Investor is treated as a direct or indirect Non-Electing U.S. Holder even if we are not a PFIC for such years. A U.S. Investor is encouraged to consult its tax advisor with respect to any available elections that may be applicable in such a situation, including the “deemed sale” election of Code Section 1298(b)(1) (which will be taxed under the adverse tax rules described above). In addition, U.S. Investors should consult their tax advisors regarding the IRS information reporting and filing obligations that may arise as a result of the ownership of shares in a PFIC.
We may invest in the equity of foreign corporations that are PFICs or may own subsidiaries that own PFICs. If we are classified as a PFIC, under attribution rules U.S. Investors will be subject to the PFIC rules with respect to their indirect ownership interests in such PFICs, such that a disposition of the shares of the PFIC or receipt by us of a distribution from the PFIC generally will be treated as a deemed disposition of such shares or the deemed receipt of such distribution by the U.S. Investor, subject to taxation under the PFIC rules. There can be no assurance that a U.S. Investor will be able to make a QEF election or a mark-to-market election with respect to PFICs in which we invest. Each U.S. Investor is encouraged to consult its own tax advisor with respect to tax consequences of an investment by us in a corporation that is a PFIC.
The U.S. federal income tax rules relating to PFICs, QEF elections, and mark-to market elections are complex. U.S. Investors are urged to consult their own tax advisors with respect to the purchase, ownership and disposition of our shares or ADSs , any elections available with respect to such shares or ADSs and the IRS information reporting obligations with respect to the purchase, ownership and disposition of our shares or ADSs .
Certain Reporting Requirements
Certain U.S. Investors are required to file IRS Form 926, Return by U.S. Transferor of Property to a Foreign Corporation, and certain U.S. Investors may be required to file IRS Form 5471, Information Return of U.S. Persons With Respect to Certain Foreign Corporations, reporting transfers of cash or other property to us and information relating to the U.S. Investor and us. Substantial penalties may be imposed upon a U.S. Investor that fails to comply.
In addition, recently enacted legislation requires certain U.S. Investors to report information on IRS Form 8938 with respect to their investments in certain “foreign financial assets,” which under certain circumstances would include an investment in our shares and ADSs, to the IRS.
Investors who fail to report required information could become subject to substantial penalties. U.S. Investors should consult their tax advisors regarding the possible implications of these reporting requirements on their investment in our shares and ADSs.
Backup Withholding Tax and Information Reporting Requirements
Generally, information reporting requirements will apply to distributions on our shares or ADSs or proceeds on the disposition of our shares or ADSs paid within the United States (and, in certain cases, outside the United States) to U.S. Investors other than certain exempt recipients, such as corporations. Furthermore, backup withholding (currently at 28%) may apply to such amounts if the U.S. Investor fails to (i) provide a correct taxpayer identification number, (ii) report interest and dividends required to be shown on its U.S. federal income tax return, or (iii) make other appropriate certifications in the required manner. U.S. Investors who are required to establish their exempt status generally must provide such certification on IRS Form W-9.
Backup withholding is not an additional tax. Amounts withheld as backup withholding from a payment may be credited against a U.S. Investor's U.S. federal income tax liability and such U.S. Investor may obtain a refund of any excess amounts withheld by filing the appropriate claim for refund with the IRS and furnishing any required information in a timely manner.
| 113 |
New Legislative Developments
With respect to taxable years beginning after December 31, 2012, certain U.S. persons, including individuals, estates and trusts, will be subject to an additional 3.8% Medicare tax on unearned income. For individuals, the additional Medicare tax applies to the lesser of (i) “net investment income” or (ii) the excess of “modified adjusted gross income” over $200,000 ($250,000 if married and filing jointly or $125,000 if married and filing separately). “Net investment income” generally equals the taxpayer’s gross investment income reduced by the deductions that are allocable to such income. Investment income generally includes passive income such as interest, dividends, annuities, royalties, rents, and capital gains. U.S. Investors are urged to consult their own tax advisors regarding the implications of the additional Medicare tax resulting from their ownership and disposition of our shares or ADSs .
U.S. Investors should consult their own tax advisors concerning the tax consequences relating to the purchase, ownership and disposition of our shares or ADSs .
F. Dividends and Paying Agents.
Not applicable.
G. Statements by Experts.
Not applicable.
H. Documents on Display.
We are subject to the information reporting requirements of the Exchange Act, applicable to foreign private issuers and under those requirements will file reports with the SEC. Those other reports or other information and this Annual Report may be inspected without charge at 10 Bareket Street, Kiryat Matalon, Petah-Tikva 4951778, Israel, and inspected and copied at the public reference facilities of the SEC located at 100 F Street, N.E., Washington, D.C. 20549. You may also obtain copies of the documents at prescribed rates by writing to the Public Reference Section of the SEC at 100 F Street, N.E., Washington, DC 20549. Please call the SEC at 1-800-SEC-0330 for further information on the public reference room. The SEC also maintains a website at http://www.sec.gov from which certain filings may be accessed.
As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file annual, quarterly and current reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. However, we will file with the SEC, within 120 days after the end of each fiscal year, or such applicable time as required by the SEC, an annual report on Form 20-F containing financial statements audited by an independent registered public accounting firm, and will submit to the SEC, on a Form 6-K, unaudited quarterly financial information.
In addition, because our ordinary shares are traded on the TASE, we have filed Hebrew language periodic and immediate reports with, and furnish information to, the TASE and the ISA, as required under Chapter Six of the Israel Securities Law. Copies of our filings with the ISA can be retrieved electronically through the MAGNA distribution site of the ISA (www.magna.isa.gov.il) and the TASE website (www.maya.tase.co.il).
We maintain a corporate website at www.canfite.com. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report on Form 20-F. We have included our website address in this Annual Report on Form 20-F solely as an inactive textual reference.
I. Subsidiary Information.
Not applicable.
ITEM 11. Quantitative and Qualitative Disclosures About Market Risk
Market risk is the risk of loss related to changes in market prices, including interest rates and foreign exchange rates, of financial instruments that may adversely impact our consolidated financial position, results of operations or cash flows.
Interest Rate Risk
We do not anticipate undertaking any significant long-term borrowings. At present, our investments consist primarily of cash and cash equivalents. We may invest in investment-grade marketable securities with maturities of up to three years, including commercial paper, money market funds, and government/non-government debt securities. The primary objective of our investment activities is to preserve principal while maximizing the income that we receive from our investments without significantly increasing risk and loss. Our investments are exposed to market risk due to fluctuation in interest rates, which may affect our interest income and the fair market value of our investments, if any. We manage this exposure by performing ongoing evaluations of our investments. Due to the short-term maturities, if any, of our investments to date, their carrying value has always approximated their fair value. If we decide to invest in investments other than cash and cash equivalents, it will be our policy to hold such investments to maturity in order to limit our exposure to interest rate fluctuations.
| 114 |
Foreign Currency Exchange Risk
Our foreign currency exposures give rise to market risk associated with exchange rate movements of the NIS, our functional and reporting currency, mainly against the dollar and the euro. Although the NIS is our functional currency, a significant portion of our expenses are denominated in both dollars and Euros and currently all of our revenues are denominated in dollars. Our U.S. dollar and euro expenses consist principally of payments made to sub-contractors and consultants for preclinical studies, clinical trials and other research and development activities. We anticipate that a sizable portion of our expenses will continue to be denominated in currencies other than the NIS. If the NIS fluctuates significantly against either the U.S. dollar or the euro, it may have a negative impact on our results of operations. To date, fluctuations in the exchange rates have not materially affected our results of operations or financial condition for the periods under review.
To date, we have not engaged in hedging transactions. In the future, we may enter into currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rates of our principal operating currencies. These measures, however, may not adequately protect us from the material adverse effects of such fluctuations.
ITEM 12. Description of Securities Other Than Equity Securities
A. Debt Securities.
Not applicable.
B. Warrants and Rights.
Not applicable.
C. Other Securities.
Not applicable.
D. American Depositary Shares
The Bank of New York Mellon, as Depositary, has registered and delivered American Depositary Shares, or ADSs. Each ADS represents two (2) ordinary shares (or a right to receive two (2) ordinary shares) deposited with the principal Tel Aviv office of Bank Hapoalim, as custodian for the Depositary. Each ADS also represents any other securities, cash or other property which may be held by the Depositary. The Depositary’s corporate trust office at which the ADSs are administered is located at 101 Barclay Street, New York, New York 10286. The Bank of New York Mellon’s principal executive office is located at One Wall Street, New York, New York 10286.
The form of the deposit agreement for the ADSs and the form of American Depositary Receipt that represents an ADS have been incorporated by reference as exhibits to this Annual Report on Form 20-F.
| 115 |
Fees and Expenses
| Persons depositing or withdrawing shares orADS holders must pay: | For: | ||
| $5.00 (or less) per 100 ADSs (or portion of 100 ADSs) | ● | Issuance of ADSs, including issuances resulting from a distribution of shares or rights or other property | |
| ● | Cancellation of ADSs for the purpose of withdrawal, including if the Deposit Agreement terminates | ||
| $.05 (or less) per ADS | ● | Any cash distribution to ADS holders | |
| A fee equivalent to the fee that would be payable if securities distributed to you had been shares and the shares had been deposited for issuance of ADSs | ● | Distribution of securities distributed to holders of deposited securities which are distributed by the Depositary to ADS holders | |
| $.05 (or less) per ADSs per calendar year | ● | Depositary services | |
| Registration or transfer fees | ● | Transfer and registration of shares on our share register to or from the name of the Depositary or its agent when you deposit or withdraw shares | |
| Expenses of the Depositary | ● | Cable, telex and facsimile transmissions (when expressly provided in the Deposit Agreement) | |
| ● | Converting foreign currency to U.S. dollars | ||
| Taxes and other governmental charges the Depositary or the custodian have to pay on any ADS or share underlying an ADS, for example, stock transfer taxes, stamp duty or withholding taxes | ● | As necessary | |
| Any charges incurred by the Depositary or its agents for servicing the deposited securities | ● | As necessary | |
The Depositary collects its fees for delivery and surrender of ADSs directly from investors depositing shares or surrendering ADSs for the purpose of withdrawal or from intermediaries acting for them. The Depositary collects fees for making distributions to investors by deducting those fees from the amounts distributed or by selling a portion of distributable property to pay the fees. The Depositary may collect its annual fee for depositary services by deduction from cash distributions, by directly billing investors or by charging the book-entry system accounts of participants acting for them. The Depositary may generally refuse to provide fee-attracting services until its fees for those services are paid.
From time to time, the Depositary may make payments to us to reimburse us for expenses and/or share revenue with us from the fees collected from ADS holders, or waive fees and expenses for services provided, generally relating to costs and expenses arising out of the establishment and maintenance of the ADS program. In performing its duties under the Deposit Agreement, the Depositary may use brokers, dealers or other service providers that are affiliates of the Depositary and that may earn or share fees or commissions.
| 116 |
ITEM 13. Defaults, Dividend Arrearages and Delinquencies
Not applicable.
ITEM 14. Material Modifications to the Rights of Security Holders and Use of Proceeds
Registered Direct Offering
The effective date of the registration statement, File No. 333-199033, on Form F-3 was October 21, 2014. In a registered direct offering that closed on December 8, 2014, we sold an aggregate of 1,797,753 ADSs at $4.45 per share for aggregate gross proceeds of $8,000,000. In addition, the investors received unregistered warrants to purchase 898,877 ADSs. The warrants may be exercised at any time for a period of five years from issuance and have an exercise price of $4.45 per ADS, subject to adjustment as set forth therein. The warrants may be exercised on a cashless basis if six months after issuance there is no effective registration statement registering the ADSs underlying the warrants.
H.C. Wainwright & Co., LLC acted as placement agent and Roth Capital Partners LLC acted as a financial advisor in connection with the offering.
The total expenses of the offering, including placement agent fees were approximately $0.8 million. The net proceeds that we received from the offering were approximately $7.2 million.
From the closing until December 31, 2016, we have used the entire net proceeds from the offering for research and development expenses, working capital and other general corporate purposes.
None of the net proceeds of the offering was paid directly or indirectly to any director, officer, general partner of ours or to their associates, persons owning ten percent or more of any class of our equity securities, or to any of our affiliates, except as employee/consultant compensation and general and administrative expenses.
ITEM 15. Controls and Procedures
Disclosure controls and procedures
Our management, including our chief executive officer, or CEO, and our chief financial officer, or CFO, are responsible for establishing and maintaining our disclosure controls and procedures (within the meaning of Rule 13a-15(e) of the Exchange Act). These controls and procedures were designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC and that such information is accumulated and communicated to our management, including our CEO and CFO, as appropriate, to allow timely decisions regarding required disclosure. We evaluated these disclosure controls and procedures under the supervision of our CEO and CFO as of December 31, 2016. Based upon that evaluation, our management, including our CEO and CFO, concluded that our disclosure controls and procedures as of December 31, 2016 were effective.
Management's annual report on internal control over financial reporting
Our management, including our CEO, and our CFO, are responsible for establishing and maintaining adequate internal control over our financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act of 1934, as amended. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Internal control over financial reporting includes policies and procedures that:
| ● | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect our transactions and asset dispositions; | |
| ● | provide reasonable assurance that transactions are recorded as necessary to permit the preparation of our financial statements in accordance with generally accepted accounting principles; | |
| ● | provide reasonable assurance that receipts and expenditures are made only in accordance with 370authorizations of our management and board of directors (as appropriate); and | |
| ● | provide reasonable assurance regarding the prevention or timely detection of unauthorized acquisition, use or disposition of assets that could have a material effect on our financial statements. |
| 117 |
Due to its inherent limitations, internal controls over financial reporting may not prevent or detect misstatements. In addition, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of our management, including our CEO, and our CFO, we assessed the effectiveness of our internal control over financial reporting as of December 31, 2016 based on the framework for Internal Control-Integrated Framework set forth by The Committee of Sponsoring Organizations of the Treadway Commission (COSO)(2013).
Based on our assessment and this framework, our management concluded that our internal control over financial reporting were effective as of December 31, 2016.
Attestation Report of Registered Public Accounting Firm
Not applicable.
Changes in internal control over financial reporting
There were no changes in our internal control over financial reporting, other than as described above, that occurred during the year ended December 31, 2016 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 16A. Audit Committee Financial Expert
Our Board of Directors has determined that Gil Oren is an audit committee financial expert, as defined by applicable SEC regulations. Gil Oren qualified as an "independent director," as that term is defined under NYSE MKT rules.
We have adopted a code of ethics, referred to as a Code of Business Conduct, applicable to our directors, officers and all other employees. Our code of ethics is publicly available on our website at www.canfite.com. If we make any amendment to the code of ethics or grant any waivers, including any implicit waiver, from a provision of the code of ethics, which applies to our chief executive officer, chief financial officer, chief accounting officer or controller, or persons performing similar functions, we will disclose the nature of such amendment or waiver on our website.
ITEM 16C. Principal Accountant Fees and Services
The following table sets forth, for each of the years indicated, the fees billed by our independent registered public accounting firm.
Year Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Services Rendered | (in thousands of NIS) | |||||||
| Audit (1) | 370 | 370 | ||||||
| Audit related services (2) | 23 | 19 | ||||||
| Tax | - | - | ||||||
| All other fees (3) | - | 9 | ||||||
| Total | 393 | 398 | ||||||
| (1) | Audit fees consist of services that would normally be provided in connection with statutory and regulatory filings or engagements, including services that generally only the independent accountant can reasonably provide. |
| (2) | Audit related services consist of services that were reasonably related to the performance of the audit or reviews of our financial statements and not included under “Audit Fees” above, including, principally, providing consents for registration statement filings. |
| (3) | All other fees consist of consulting services related to compensation and risk management. |
| 118 |
Audit Committee Pre-Approval Policies and Procedures
Our audit committee’s specific responsibilities in carrying out its oversight of the quality and integrity of the accounting, auditing and reporting practices of us include the approval of audit and non-audit services to be provided by the external auditor. The audit committee approves in advance the particular services or categories of services to be provided to us during the following yearly period and also sets forth a specific budget for such audit and non-audit services. Additional non-audit services may be pre-approved by the audit committee.
ITEM 16D. Exemptions from the Listing Standards for Audit Committees
Not applicable.
ITEM 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers
Not applicable.
ITEM 16F. Change in Registrant’s Certifying Accountant
Not applicable.
ITEM 16G. Corporate Governance
We are a foreign private issuer whose ordinary shares are listed on the NYSE MKT. As such, we are required to comply with U.S. federal securities laws, including the Sarbanes-Oxley Act, and the NYSE MKT rules, including the NYSE MKT corporate governance requirements. The NYSE MKT rules provide that foreign private issuers may follow home country practice in lieu of certain qualitative listing requirements subject to certain exceptions and except to the extent that such exemptions would be contrary to U.S. federal securities laws, so long as the foreign issuer discloses that it does not follow such listing requirement and describes the home country practice followed in its reports filed with the SEC. Below is a concise summary of the significant ways in which our corporate governance practices differ from the corporate governance requirements of NYSE MKT applicable to domestic U.S. listed companies:
| ● | The NYSE MKT rules recommend that an issuer have a quorum requirement for shareholders meetings of at least one-third of the outstanding shares of the issuer’s common voting stock. We have chosen to follow home country practice with respect to the quorum requirements of our shareholders meeting and our adjourned shareholders meeting. Our articles of association, as permitted under the Israeli Companies Law and Israeli practice, provide that the quorum requirements for a shareholders meeting are the presence of at least two shareholders who represent at least 25%of the outstanding shares of the issuer’s common voting stock, and in the event of an adjourned meeting, the presence of a minimum of two shareholders present in person. |
| ● | We have chosen to follow our home country practice in lieu of the requirements of the NYSE MKT rules relating to an issuer’s furnishing of its annual report to shareholders. However, we post our Annual Report on Form 20-F on our web site (www.canfite.com) as soon as practicable following the filing of the Annual Report on Form 20-F with the SEC. |
| ● | We have chosen to follow our home country practice in lieu of the requirements of the NYSE MKT rules relating to shareholder approval required prior to the issuance of securities (i) when a stock option or purchase plan is to be established or materially amended or other equity compensation arrangement made or materially amended, pursuant to which stock may be acquired by officers, directors, employees or consultants and (ii) in connection with a transaction, other than a public offering, involving the issuance or potential issuance by the Company of ordinary shares (or their equivalent) equal to 20% or more of the ordinary shares or 20% voting power outstanding before the issuance for or at a price less than the greater of book or market value of the shares. We follow the provisions of the Israeli Companies Law with regard to transactions with our affiliates, i.e., our controlling shareholder and our directors and officers, including private placement transactions. |
ITEM 16H. Mine Safety Disclosure
Not applicable.
| 119 |
We have responded to Item 18 in lieu of responding to this item.
Please refer to the financial statements beginning on page F-1.
| Page | ||||
| Report of Independent Registered Public Accounting Firm | F-2 | |||
| Audited Consolidated Financial Statements as of December 31, 2016 and 2015 and for each of the three years in the period ended December 31, 2016 | ||||
| Consolidated Statements of Financial Position | F-3 | |||
| Consolidated Statements of Comprehensive Loss | F-5 | |||
| Consolidated Statements of Changes in Equity (Deficiency) | F-8 | |||
| Consolidated Statements of Cash Flows | F-10 | |||
| Notes to Consolidated Financial Statements | F-12 |
The following financial statements and financial statement schedules are filed as part of this Annual Report on Form 20-F, together with the report of the independent registered public accounting firm.
| 120 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
CONSOLIDATED FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2016
INDEX
- - - - - - - - - - -
| F-1 |
 |
Kost Forer Gabbay & Kasierer 3 Aminadav St. Tel-Aviv 6706703, Israel
|
Tel: +972-3-6232525 Fax: +972-3-5622555 ey.com |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
We have audited the accompanying consolidated statements of financial position of Can-Fite Biopharma Ltd. and its subsidiaries (the “Company”) as of December 31, 2015 and 2016, and the related consolidated statements of comprehensive loss, changes in equity and cash flows for each of the three years in the period ended December 31, 2016. These financial statements are the responsibility of the Company's board of directors and management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company's and its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements,. assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, based on our audits, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of the Company and its subsidiaries as of December 31, 2015 and 2016, and consolidated statements of comprehensive loss, changes in equity and cash flows for each of the three years in the period ended December 31, 2016, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
| Tel-Aviv, Israel | KOST FORER GABBAY & KASIERER | |
| March 30, 2017 | A Member of Ernst & Young Global |
| F-2 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
CONSOLIDATED STATEMENTS OF FINANCIAL POSITION
In thousands (except for share and per share data)
| December 31, | |||||||||||||||
| 2016 | 2016 | 2015 | |||||||||||||
| Note | USD | NIS | |||||||||||||
| Note 2.c.2 | |||||||||||||||
| ASSETS | |||||||||||||||
| CURRENT ASSETS: | |||||||||||||||
| Cash and cash equivalents | 8,115 | 31,203 | 66,026 | ||||||||||||
| Other accounts receivables and prepaid expenses | 5 | 1,993 | 7,664 | 2,419 | |||||||||||
| Total current assets | 10,108 | 38,867 | 68,445 | ||||||||||||
| NON-CURRENT ASSETS: | |||||||||||||||
| Lease deposit | 10 | 37 | 27 | ||||||||||||
| Property, plant and equipment, net | 6 | 53 | 205 | 236 | |||||||||||
| Total long-term assets | 63 | 242 | 263 | ||||||||||||
| Total assets | 10,171 | 39,109 | 68,708 | ||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-3 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
CONSOLIDATED STATEMENTS OF FINANCIAL POSITION
In thousands (except for share and per share data)
| December 31, | |||||||||||||||
| 2016 | 2016 | 2015 | |||||||||||||
| Note | USD | NIS | |||||||||||||
| Note 2.c.2 | |||||||||||||||
| LIABILITIES AND SHAREHOLDERS' EQUITY | |||||||||||||||
| CURRENT LIABILITIES: | |||||||||||||||
| Trade payables | 1,249 | 4,804 | 1,803 | ||||||||||||
| Deferred revenues | 10 | 322 | 1,237 | 857 | |||||||||||
| Other accounts payable | 7 | 933 | 3,588 | 4,279 | |||||||||||
| Total current liabilities | 2,504 | 9,629 | 6,939 | ||||||||||||
| NON-CURRENT LIABILITIES: | |||||||||||||||
| Warrants exercisable into shares | 8 | 2,618 | 10,068 | 16,725 | |||||||||||
| Deferred revenues | 10 | 1,173 | 4,510 | 3,641 | |||||||||||
| Severance pay, net | 9 | - | - | 630 | |||||||||||
| Total Long-term liabilities | 3,791 | 14,578 | 20,996 | ||||||||||||
| CONTIGENT LIABILITIES AND COMMITMENTS | 10 | ||||||||||||||
| EQUITY ATTRIBUTABLE TO EQUITY HOLDERS OF THE COMPANY: | 11 | ||||||||||||||
| Share capital | 1,831 | 7,039 | 7,030 | ||||||||||||
| Share premium | 86,573 | 332,873 | 332,873 | ||||||||||||
| Capital reserve from share-based payment transactions | 5,315 | 20,438 | 19,288 | ||||||||||||
| Warrants exercisable into shares (Series 10-12) | 2,336 | 8,983 | 8,983 | ||||||||||||
| Treasury shares, at cost | (943 | ) | (3,628 | ) | (3,628 | ) | |||||||||
| Accumulated other comprehensive loss | (229 | ) | (883 | ) | (1,401 | ) | |||||||||
| Accumulated deficit | (91,015 | ) | (349,953 | ) | (322,876 | ) | |||||||||
| Total equity attributable to equity holders of the company | 3,868 | 14,869 | 40,269 | ||||||||||||
| Non-controlling interests | 8 | 33 | 504 | ||||||||||||
| Total equity | 3,876 | 14,902 | 40,773 | ||||||||||||
| Total liabilities and equity | 10,171 | 39,109 | 68,708 | ||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-4 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
In thousands (except for share and per share data)
| Year ended December 31, | |||||||||||||||||||
| 2016 | 2016 | 2015 | 2014 | ||||||||||||||||
| Note | USD | NIS | |||||||||||||||||
| Note 2.c.2 | |||||||||||||||||||
| Revenues | 10 | 170 | 652 | 643 | - | ||||||||||||||
| Research and development expenses | 13 | 6,081 | 23,380 | 15,052 | 16,200 | ||||||||||||||
| General and administrative expenses | 14 | 2,726 | 10,483 | 10,633 | 11,573 | ||||||||||||||
| Operating loss | 8,637 | 33,211 | 25,042 | 27,773 | |||||||||||||||
| Financial expenses | 15 | 178 | 685 | 2,203 | 1,228 | ||||||||||||||
| Financial income | 15 | (1,820 | ) | (6,999 | ) | (7,492 | ) | (4,500 | ) | ||||||||||
| Loss before taxes on income | 6,995 | 26,897 | 19,753 | 24,501 | |||||||||||||||
| Taxes on income | 17 | 29 | 112 | 17 | 23 | ||||||||||||||
| Net loss | 7,024 | 27,009 | 19,770 | 24,524 | |||||||||||||||
| Other comprehensive loss: | |||||||||||||||||||
| Adjustments arising from translating financial statements of foreign operations | 9 | 33 | 1 | 939 | |||||||||||||||
| Remeasurement loss from defined benefit plans | - | - | 385 | 94 | |||||||||||||||
| Total other comprehensive loss | 9 | 33 | 386 | 1,033 | |||||||||||||||
| Total comprehensive loss | 7,033 | 27,042 | 20,156 | 25,557 | |||||||||||||||
| Net loss Attributable to: | |||||||||||||||||||
| Equity holders of the Company | 6,900 | 26,532 | 18,726 | 23,759 | |||||||||||||||
| Non-controlling interests | 124 | 477 | 1,044 | 765 | |||||||||||||||
| 7,024 | 27,009 | 19,770 | 24,524 | ||||||||||||||||
| Total comprehensive loss attributable to: | |||||||||||||||||||
| Equity holders of the Company | 6,907 | 26,559 | 19,112 | 24,623 | |||||||||||||||
| Non-controlling interests | 126 | 483 | 1,044 | 934 | |||||||||||||||
| 7,033 | 27,042 | 20,156 | 25,557 | ||||||||||||||||
| Net loss per share attributable to equity holders of the Company: | |||||||||||||||||||
| Basic and diluted net loss per share | 16 | 0.25 | 0.96 | 0.81 | 1.35 | ||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-5 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
NIS in thousands (except for share and per share data)
| Attributable to equity holders of the Company | ||||||||||||||||||||||||||||||||||||||||
Share capital | Share premium | Capital reserve from share-based payment transactions |
Warrants exercisable into shares | Treasury shares | Accumulated other comprehensive income (loss) | Accumulated deficit | Total | Non-controlling interests | Total Equity | |||||||||||||||||||||||||||||||
| Balance as of January 1, 2014 | 4,037 | 267,946 | 15,761 | 9,652 | (3,628 | ) | (151 | ) | (280,391 | ) | 13,226 | 2,299 | 15,525 | |||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (23,759 | ) | (23,759 | ) | (765 | ) | (24,524 | ) | ||||||||||||||||||||||||||
| Adjustments arising from translating financial statements of foreign operations | - | - | - | - | - | (770 | ) | - | (770 | ) | (169 | ) | (939 | ) | ||||||||||||||||||||||||||
| Remeasurement gain (loss) from defined benefit plans | - | - | - | - | - | (94 | ) | - | (94 | ) | - | (94 | ) | |||||||||||||||||||||||||||
| Total comprehensive loss | - | - | - | - | - | (864 | ) | (23,759 | ) | (24,623 | ) | (934 | ) | (25,557 | ) | |||||||||||||||||||||||||
| Issuance of share capital and warrants, net of issue expenses of NIS 3,845 | 1,390 | 33,522 | 994 | - | - | - | - | 35,906 | - | 35,906 | ||||||||||||||||||||||||||||||
| Share-based payments | 14 | 319 | 398 | - | - | - | - | 731 | 95 | 826 | ||||||||||||||||||||||||||||||
| Exercise of unlisted share options | *)- | *)- | - | - | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
| Balance as of December 31, 2014 | 5,441 | 301,787 | 17,153 | 9,652 | (3,628 | ) | (1,015 | ) | (304,150 | ) | 25,240 | 1,460 | 26,700 | |||||||||||||||||||||||||||
| *) | Represent an amount lower than NIS 1. |
The accompanying notes are an integral part of the consolidated financial statements.
| F-6 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
NIS in thousands (except for share and per share data)
| Attributable to equity holders of the Company | ||||||||||||||||||||||||||||||||||||||||
Share capital | Share premium | Capital reserve from share-based payment transactions |
Warrants exercisable into shares | Treasury shares | Accumulated other comprehensive income (loss) | Accumulated deficit | Total | Non-controlling interests | Total Equity | |||||||||||||||||||||||||||||||
| Balance as of January 1, 2015 | 5,441 | 301,787 | 17,153 | 9,652 | (3,628 | ) | (1,015 | ) | (304,150 | ) | 25,240 | 1,460 | 26,700 | |||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (18,726 | ) | (18,726 | ) | (1,044 | ) | (19,770 | ) | ||||||||||||||||||||||||||
| Adjustments arising from translating financial statements of foreign operations | - | - | - | - | - | (1 | ) | - | (1 | ) | - | (1 | ) | |||||||||||||||||||||||||||
| Remeasurement gain (loss) from defined benefit plans | - | - | - | - | - | (385 | ) | - | (385 | ) | - | (385 | ) | |||||||||||||||||||||||||||
| Total comprehensive loss | - | - | - | - | - | (386 | ) | (18,726 | ) | (19,112 | ) | (1,044 | ) | (20,156 | ) | |||||||||||||||||||||||||
| Issuance of share capital and warrants, net of issue expenses of NIS 3,530 | 1,589 | 30,417 | 1,781 | - | - | - | - | 33,787 | - | 33,787 | ||||||||||||||||||||||||||||||
| Expiration of warrants exercisable into shares | - | 669 | - | (669 | ) | - | - | - | - | - | - | |||||||||||||||||||||||||||||
| Share-based payments | - | - | 354 | - | - | - | - | 354 | 88 | 442 | ||||||||||||||||||||||||||||||
| Balance as of December 31, 2015 | 7,030 | 332,873 | 19,288 | 8,983 | (3,628 | ) | (1,401 | ) | (322,876 | ) | 40,269 | 504 | 40,773 | |||||||||||||||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-7 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
NIS in thousands (except for share and per share data)
| Attributable to equity holders of the Company | ||||||||||||||||||||||||||||||||||||||||
Share capital | Share premium | Capital reserve from share-based payment transactions |
Warrants exercisable into shares | Treasury shares | Accumulated other comprehensive income (loss) | Accumulated deficit | Total | Non-controlling interests | Total Equity | |||||||||||||||||||||||||||||||
| Balance as of January 1, 2016 | 7,030 | 332,873 | 19,288 | 8,983 | (3,628 | ) | (1,401 | ) | (322,876 | ) | 40,269 | 504 | 40,773 | |||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (26,532 | ) | (26,532 | ) | (477 | ) | (27,009 | ) | ||||||||||||||||||||||||||
| Adjustments arising from translating financial statements of foreign operations | - | - | - | - | - | (27 | ) | - | (27 | ) | (6 | ) | (33 | ) | ||||||||||||||||||||||||||
| Loss from defined benefit plans | - | - | - | - | - | 545 | (545 | ) | - | - | - | |||||||||||||||||||||||||||||
| Total comprehensive loss | - | - | - | - | - | 518 | (27,077 | ) | (26,559 | ) | (483 | ) | (27,042 | ) | ||||||||||||||||||||||||||
| Share-based payments | 9 | - | 1,150 | - | - | - | - | 1,159 | 12 | 1,171 | ||||||||||||||||||||||||||||||
| Balance as of December 31, 2016 | 7,039 | 332,873 | 20,438 | 8,983 | (3,628 | ) | (883 | ) | (349,953 | ) | 14,869 | 33 | 14,902 | |||||||||||||||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-8 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
In thousands (except for share and per share data)
| Attributable to equity holders of the Company | ||||||||||||||||||||||||||||||||||||||||
Share capital | Share Premium | Capital reserve from share-based payment transactions | Warrants exercisable into shares | Treasury shares | Accumulated other comprehensive income (loss) | Accumulated deficit | Total | Non-controlling interests | Total Equity | |||||||||||||||||||||||||||||||
| USD (Note 2.c.2) | ||||||||||||||||||||||||||||||||||||||||
| Balance as of January 1, 2016 | 1,828 | 86,573 | 5,016 | 2,336 | (943 | ) | (364 | ) | (83,973 | ) | 10,473 | 131 | 10,604 | |||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (6,900 | ) | (6,900 | ) | (124 | ) | (7,024 | ) | ||||||||||||||||||||||||||
| Adjustments arising from translating financial statements of foreign operations | (7 | ) | - | (7 | ) | (2 | ) | (9 | ) | |||||||||||||||||||||||||||||||
| Loss from defined benefit plans | - | - | - | - | - | 142 | (142 | ) | - | - | - | |||||||||||||||||||||||||||||
| - | - | - | - | - | 135 | (7,042 | ) | (6,907 | ) | (126 | ) | (7,033 | ) | |||||||||||||||||||||||||||
| Total comprehensive loss | ||||||||||||||||||||||||||||||||||||||||
| Share-based payments | 3 | - | 299 | - | - | - | - | 302 | 3 | 305 | ||||||||||||||||||||||||||||||
| Balance as of December 31, 2016 | 1,831 | 86,573 | 5,315 | 2,336 | (943 | ) | (229 | ) | (91,015 | ) | 3,868 | 8 | 3,876 | |||||||||||||||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-9 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
In thousands (except for share and per share data)
| Year ended December 31, | ||||||||||||||||
| 2016 | 2016 | 2015 | 2014 | |||||||||||||
| USD | NIS | |||||||||||||||
| Note 2.c.2 | ||||||||||||||||
| Cash flows from operating activities: | ||||||||||||||||
| Net loss | (7,024 | ) | (27,009 | ) | (19,770 | ) | (24,524 | ) | ||||||||
| Adjustments to reconcile loss to net cash used: | ||||||||||||||||
| Depreciation of property, plant and equipment | 18 | 71 | 64 | 47 | ||||||||||||
| Share-based payment | 305 | 1,171 | 442 | 826 | ||||||||||||
| Issuance expenses related to warrants exercisable into shares | - | - | 2,122 | 1,170 | ||||||||||||
| Decrease in severance pay, net | (164 | ) | (630 | ) | 21 | 1 | ||||||||||
| Changes in fair value of warrants liability exercisable into shares | (1,732 | ) | (6,657 | ) | (6,913 | ) | (3,089 | ) | ||||||||
| Exchange differences on balances of cash and cash equivalents | 116 | 443 | 73 | 782 | ||||||||||||
| (1,457 | ) | (5,602 | ) | (4,191 | ) | (263 | ) | |||||||||
| Working capital adjustments: | ||||||||||||||||
| Decrease (increase) in accounts receivable, prepaid expenses and lease deposit | (1,367 | ) | (5,255 | ) | 996 | (1,181 | ) | |||||||||
| Increase (decrease) in trade payable | 756 | 2,908 | 779 | (1,069 | ) | |||||||||||
| Increase in deferred revenues | 324 | 1,249 | 4,498 | - | ||||||||||||
| Increase (decrease) in other accounts payable | (164 | ) | (631 | ) | (471 | ) | (1,495 | ) | ||||||||
| (450 | ) | (1,729 | ) | 5,802 | (3,745 | ) | ||||||||||
| Net cash used in operating activities | (8,931 | ) | (34,340 | ) | (18,159 | ) | (28,532 | ) | ||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-10 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
In thousands (except for share and per share data)
| Year ended December 31, | ||||||||||||||||
| 2016 | 2016 | 2015 | 2014 | |||||||||||||
| USD | NIS | |||||||||||||||
| Note 2.c.2 | ||||||||||||||||
| Cash flows from investing activities: | ||||||||||||||||
| Purchase of property, plant and equipment | (10 | ) | (40 | ) | (177 | ) | (37 | ) | ||||||||
| Proceeds from sale of property, plant and equipment | - | - | 10 | - | ||||||||||||
| Net cash provided by (used in) investing activities | (10 | ) | (40 | ) | (167 | ) | (37 | ) | ||||||||
| Cash flows from financing activities: | ||||||||||||||||
| Issuance of share capital and warrants, net of issuance expenses | - | - | 48,334 | 44,675 | ||||||||||||
| Net cash provided by financing activities | - | - | 48,334 | 44,675 | ||||||||||||
| Exchange differences on balances of cash and cash equivalents | (116 | ) | (443 | ) | (73 | ) | (782 | ) | ||||||||
| Increase (decrease) in cash and cash equivalents | (9,057 | ) | (34,823 | ) | 29,935 | 15,324 | ||||||||||
| Cash and cash equivalents at the beginning of the year | 17,172 | 66,026 | 36,091 | 20,767 | ||||||||||||
| Cash and cash equivalents at the end of the year | 8,115 | 31,203 | 66,026 | 36,091 | ||||||||||||
| Supplemental disclosure of cash flow information: | ||||||||||||||||
| Cash paid during the year for income taxes | 29 | 112 | 16 | 9 | ||||||||||||
| Cash received during the year for interest | 44 | 168 | 41 | 42 | ||||||||||||
| *) | Represent an amount lower than NIS 1. |
The accompanying notes are an integral part of the consolidated financial statements.
| F-11 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1:- GENERAL
| a. | Company description: |
Can-Fite Biopharma Ltd. (the “Company”) was incorporated and started to operate in September 1994 as a private Israeli company. Can-Fite is a clinical-stage biopharmaceutical company focused on developing orally bioavailable small molecule therapeutic products for the treatment of autoimmune-inflammatory indications, oncology and liver diseases as well as sexual dysfuction. Its platform technology utilizes the Gi protein associated A3AR as a therapeutic target. A3AR is highly expressed in inflammatory and cancer cells, and not significantly expressed in normal cells, suggesting that the receptor could be a unique target for pharmacological intervention. The Company's pipeline of drug candidates are synthetic, highly specific agonists and allosteric modulators, or ligands or molecules that initiate molecular events when binding with target proteins, targeting the A3AR.
The Company’s ordinary shares have been publicly trading traded on the Tel-Aviv Stock Exchange since October 2005 under the symbol “CFBI” and the Company’s American Depositary Shares (“ADSs”) began public trading on the over the counter market in the U.S. in October 2012 and since November 2013 the Company’s ADSs have been publicly traded on the NYSE MKT under the symbol “CANF”.
| b. | The Company owns 82% of a U.S. based subsidiary, OphthaliX Inc., (“OphthaliX”) which was developing the CF101 drug for treatment of ophthalmic indications under license from the Company. The license to develop this drug was transferred from the Company to OphthaliX in the context of an ophthalmic activity spinoff transaction. OphthaliX is traded in the over the counter market in the U.S. under the symbol “OPLI”. OphthaliX has ceased all research and development operations. |
The Company is also the sole owner of Ultratrend Limited, a private, inactive company incorporated in 2005 in the UK.
| c. | During the year ended December 31, 2016, the Company incurred net losses of NIS 27,009 thousand and it had negative cash flows from operating activities in the amount of NIS 34,340 thousand as well as accumulated losses from previous years. For further information regarding cash flow, please refer to Note 19. |
Furthermore, the Company intends to continue to finance its operating activities by raising capital and seeking collaborations with multinational companies in the industry. There are no assurances that the Company will be successful in obtaining an adequate level of financing needed for its long-term research and development activities. If the Company will not have sufficient liquidity resources, the Company may not be able to continue the development of all of its products or may be required to delay part of its development programs. The Company's management and board of directors are of the opinion that its current financial resources will be sufficient to continue the development of the Company's products at least for twelve months from the balance sheet date.
| F-12 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1:- GENERAL (Cont.)
| d. | Definitions: |
In these consolidated financial statements:
| The Company | - | Can-Fite Biopharma Ltd. | |||
| The Group | - | The Company and its subsidiaries (as defined below) | |||
| Subsidiaries | - | Companies that are controlled by the Company (as defined in IAS 27 (2008) and whose accounts are consolidated with those of the Company | |||
| OphthaliX | - | OphthaliX Inc | |||
| Eye-Fite | - | Eye-Fite Ltd. (OphthaliX Inc.’s wholly owned subsidiary) | |||
| Related parties | - | As defined in IAS 24 | |||
| NIS | - | New Israeli Shekel | |||
| USD | - | U.S. dollar | |||
| € | - | European Union Euro | |||
| CAD | - | Canadian dollar | |||
| ADS | - | American Depositary Share ("ADS"). Each ADS represents 2 ordinary shares of the Company |
NOTE 2:- SIGNIFICANT ACCOUNTING POLICIES
The following accounting policies have been applied consistently in the financial statements for all periods presented, unless otherwise stated.
| a. | Basis of presentation of the financial statements |
These financial statements have been prepared in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board. The Company's financial statements have been prepared on a cost basis, except for financial assets and liabilities (including warrants) which are presented at fair value through statement of comprehensive loss.
The preparation of the financial statements requires management to make critical accounting estimates as well as exercise judgment in the process of adopting significant accounting policies. The matters which required the exercise of significant judgment and the use of estimates, which have a material effect on amounts recognized in the financial statements, are specified in Note 3.
| b. | Consolidated financial statements |
The consolidated financial statements comprise the financial statements of companies that are controlled by the Company (i.e., subsidiaries). Control is achieved when the Company is exposed, or has the rights, to variable returns from its involvement with the investee and has the ability to affect those returns through its power over the investee. The effect of potential voting rights that are exercisable at the end of the reporting period is considered when assessing whether an entity has control. The consolidation of the financial statements commences on the date on which control is obtained and ends when such control ceases.
| F-13 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2:- SIGNIFICANT ACCOUNTING POLICIES (Cont.)
The financial statements of the Company and of the subsidiaries are prepared as of the same dates and periods. The consolidated financial statements are prepared using uniform accounting policies by all companies in the Group. Significant intragroup balances and transactions and gains or losses resulting from intragroup transactions are eliminated in full in the consolidated financial statements.
Non-controlling interests of subsidiaries represent the non-controlling shareholders' share of the total comprehensive loss of the subsidiaries and their share of the net assets. The non-controlling interests are presented in equity separately from the equity attributable to the equity holders of the Company. Losses are attributed to non-controlling interests even if they result in a negative balance of non-controlling interests in the consolidated statement of financial position.
| c. | Functional currency, presentation currency and foreign currency: |
| 1. | Functional currency and presentation currency: |
The functional currency of the Company and presentation currency of the financial statements is the NIS.
When a subsidiary’s functional currency differs from the Company's functional currency, the subsidiary financial statements are translated into the Company's functional currency so that they can be included in the consolidated financial statements.
Assets and liabilities are translated at the closing rate at the end of each reporting period.
Comprehensive loss items are translated at average exchange rates for all the relevant periods. All resulting translation differences are recognized as a separate component of other comprehensive loss in equity under “adjustments arising from translating financial statements”.
| 2. | Convenience translation: |
For the convenience of the reader, the reported NIS amounts as of December 31, 2016 have been translated into U.S. dollars, at the representative rate of exchange on December 31, 2016 (U.S. $ 1 = NIS 3.845). The U.S. dollar amounts presented in these financial statements should not be construed as representing amounts that are receivable or payable in dollars or convertible into U.S. dollars, unless otherwise indicated. The U.S. dollar amounts were rounded to whole numbers for convenience.
| F-14 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2:- SIGNIFICANT ACCOUNTING POLICIES (Cont.)
| 3. | Transactions, assets and liabilities in foreign currency: |
Transactions denominated in foreign currency are recorded upon initial recognition at the exchange rate at the date of the transaction. After initial recognition, monetary assets and liabilities denominated in foreign currency are translated at the end of each reporting period into the functional currency at the exchange rate at that date. Exchange rate differences are recognized in statement of comprehensive loss. Non-monetary assets and liabilities measured at cost in foreign currency are translated at the exchange rate at the date of the transaction. Non-monetary assets and liabilities denominated in foreign currency and measured at fair value are translated into the functional currency using the exchange rate prevailing at the date when the fair value was determined.
| 4. | Index-linked monetary items: |
Monetary assets and liabilities linked to the changes in the Israeli Consumer Price Index (“Israeli CPI”) are adjusted at the relevant index at the end of each reporting period according to the terms of the agreement. Linkage differences arising from the adjustment, as above, are recognized in statement of comprehensive loss.
| d. | Cash equivalents |
Cash equivalents are considered as highly liquid investments, including unrestricted short-term bank deposits with an original maturity of three months or less from the investment date.
| e. | Account receivables and prepaid expenses |
Prepaid expenses are composed mainly from active pharmaceutical ingredients and clinical trial drug-kits which are expensed based on the percentage of completion method of the related clinical trials.
| f. | Property, plant and equipment |
Property, plant and equipment are measured at cost, including directly attributable costs, less accumulated depreciation, accumulated impairment losses and excluding day-to-day servicing expenses.
Depreciation is calculated on a straight-line basis over the useful life of the assets at annual rates as follows:
| % | Mainly % | ||||||||
| Laboratory equipment and Leasehold improvements | 10 | 10 | |||||||
| Computers, office furniture and equipment | 6 - 33 | 33 | |||||||
Leasehold improvements are depreciated on a straight-line basis over the shorter of the lease term (including extension option held by the Company and intended to be exercised) and the expected life of the improvement.
The useful life, depreciation method and residual value of an asset are reviewed at least each year-end and any changes are accounted for prospectively as a change in accounting estimates. Depreciation of an asset ceases at the earlier of the date that the asset is classified as held for sale and the date that the asset is derecognized.
| F-15 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2:- SIGNIFICANT ACCOUNTING POLICIES (Cont.)
| g. | Revenue recognition |
The Company generates revenues from distribution agreements. Such revenues comprise of upfront license fees, milestone payments and potential royalty payments.
The Company identified four components in the agreements: (i) performing the research and development services through regulatory approval; (ii) exclusive license to distribute the product; (iii) participation in joint steering committee; and, (iv) royalties resulting from future sales of the product.
The Company recognizes revenue in accordance with IAS 18, "Revenue" pursuant to which each required deliverable is evaluated to determine whether it qualifies as a separate unit of accounting based on whether the deliverable has "stand-alone value" to the customer. The arrangement’s consideration that is fixed or determinable is then allocated to each separate unit of accounting based on the relative selling price of each deliverable which is based on the Estimated Selling Price (''ESP'').
Components (i) – (iii) were analyzed as one unit of accounting. Consequently, revenue from these components is recorded based on the term of the research and development services (which is the last deliverable in the arrangement).
Contingent payments related to milestones will be recognized immediately upon satisfaction of the milestone and contingent payments related to royalties will be recognized in the period that the related sales have occurred.
Revenues from royalties will be recognized as they accrue in accordance with the terms of the relevant agreement.
| h. | Research and development expenditures |
Research expenditures are recognized in the statement of comprehensive loss when incurred.
| i. | Impairment of non-financial assets |
The Group evaluates the need to record an impairment of the carrying amount of property, plant and equipment whenever events or changes in circumstances indicate that the carrying amount is not recoverable. If the carrying amount of property, plant and equipment exceeds their recoverable amount, the property, plant and equipment are reduced to their recoverable amount. The recoverable amount is the higher of fair value less costs of sale and value in use. In measuring value in use, the expected future cash flows are discounted using a pre-tax discount rate that reflects the risks specific to the asset. The recoverable amount of an asset that does not generate independent cash flows is determined for the cash-generating unit to which the asset belongs. Impairment losses are recognized in profit or loss. As of December 31, 2016 and 2015, no impairment losses have been identified.
| j. | Financial instruments |
| 1. | Financial liabilities |
Financial liabilities are initially recognized at fair value.
| F-16 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2:- SIGNIFICANT ACCOUNTING POLICIES (Cont.)
The Group determines the classification of the liability on the date of initial recognition. All liabilities are initially recognized at fair value. After initial recognition, the accounting treatment of financial liabilities is based on their classification as follows:
Financial liabilities at fair value through statement of comprehensive loss
Financial liabilities at fair value through profit or loss include financial liabilities designated upon initial recognition as at fair value through statement of comprehensive loss.
A liability may be designated upon initial recognition at fair value through profit or loss, subject to the provisions of IAS 39.
| 2. | Fair value |
The fair value of financial instruments that are traded in an active market is determined by reference to market prices at the end of the reporting period. For financial instruments where there is no active market, fair value is determined using valuation techniques. Such techniques include using recent arm's length market transactions, reference to the current market value of another instrument which is substantially the same, discounted cash flow and other valuation models.
A detailed analysis of the fair value measurement of financial instruments is provided in Note 8d.
| 3. | Issue of a unit of securities |
The issue of a unit of securities involves the allocation of the proceeds received (before issue expenses) to the components of the securities issued in the unit based on the following order: financial derivatives and other financial instruments measured at fair value in each period. Then fair value is determined for financial liabilities and compound instruments that are presented at amortized cost. The consideration allocated to the equity instruments is determined as the residual value. The issuance costs are allocated to each component based on the amounts allocated to each component in the unit.
| 4. | Derecognition of financial instruments |
Financial liabilities:
A financial liability is derecognized when it is extinguished, that is when the obligation is discharged, realized, cancelled or expires. A financial liability is extinguished when the debtor (i.e., the Group) discharges the liability by paying in cash, other financial assets, goods or services or shares, or is legally released from the liability.
When an existing financial liability is exchanged with another liability from the same lender on substantially different terms, or the terms of an existing liability are substantially modified, such an exchange or modification is accounted for as an extinguishment of the original liability and the recognition of a new liability. The difference between the carrying amount of the above liabilities is recognized in statement of comprehensive loss. If the exchange or modification is not substantial, it is accounted for as a change in the terms of the original liability and no gain or loss is recognized on the exchange.
| F-17 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2:- SIGNIFICANT ACCOUNTING POLICIES (Cont.)
| k. | Treasury shares |
Company shares held by OphthaliX are recognized at cost, and as a deduction from equity. Any gain or loss arising from a purchase, sale, issuance or cancellation of treasury shares is recognized directly in equity.
| l. | Provisions |
A provision in accordance with IAS 37 is recognized when the Group has a present obligation (legal or constructive) as a result of a past event, it is probable that an outflow of resources embodying economic benefits will be required to settle the obligation and a reliable estimate can be made of the amount of the obligation. If the Group expects part or all of the expense to be reimbursed to the Company, such as in an insurance contract, the reimbursement is recognized as a separate asset only when it is virtually certain that it will be received by the Company. The expense is recognized in the income statement net of the reimbursed amount.
Legal claims:
A provision for claims is recognized when the Group has a present legal or constructive obligation as a result of a past event, it is more likely than not that an outflow of resources embodying economic benefits will be required by the Group to settle the obligation and a reliable estimate can be made of the amount of the obligation. No provisions pursuant to IAS 37 have been identified.
| m. | Employee benefit liabilities |
The Group has several employee benefit plans:
| 1. | Short-term employee benefits: |
Short-term employee benefits are benefits that are expected to be settled wholly before twelve months after the end of the annual reporting period in which the employees render the related services. These benefits include salaries, paid annual leave, paid sick leave, recreation and social security contributions and are recognized as expenses as the services are rendered. A liability in respect of a cash bonus or a profit-sharing plan is recognized when the Group has a legal or constructive obligation to make such payment as a result of past service rendered by an employee and a reliable estimate of the amount can be made.
| 2. | Post-employment benefits: |
The post-employment benefit plans are normally financed by contributions to insurance companies and classified as defined benefit plans.
The Company operates a defined benefit plan in respect of severance pay pursuant to the Severance Pay Law. According to the Severance Pay Law, employees are entitled to severance pay upon dismissal or retirement. The liability for termination of employment is measured using the projected unit credit method. The actuarial assumptions include rates of employee turnover and future salary increases based on the estimated timing of payment. The amounts are presented based on discounted expected future cash flows using a discount rate determined by reference to yields on government bonds with a term that matches the estimated term of the benefit obligation.
| F-18 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2:- SIGNIFICANT ACCOUNTING POLICIES (Cont.)
In respect of its severance pay obligation to certain of its employees, the Company makes current deposits in pension funds and insurance companies (the “Plan Assets”). Plan Assets comprise assets held by a long-term employee benefit fund or qualifying insurance policies. Plan Assets are not available to the Company's own creditors and cannot be returned directly to the Company.
The liability for employee benefits shown in the statement of financial position reflects the present value of the defined benefit obligation less the fair value of the plan assets, less past service costs.
Actuarial gains and losses are recognized in other comprehensive loss.
As for effects of the change in the type of bonds used in determining the discount rate, see Note 2q.
| n. | Share-based payment transactions |
The Company's employees and other service providers are entitled to remuneration in the form of equity-settled share-based payment transactions. The cost of equity-settled transactions with employees is measured at the fair value of the equity instruments granted at grant date. The fair value is determined using the binomial option pricing model.
As for other service providers, the cost of the transactions is measured at the fair value of the goods or services received as consideration for equity instruments. In cases where the fair value of the goods or services received as consideration of equity instruments cannot be measured, they are measured by reference to the fair value of the equity instruments granted using binomial option pricing model.
The cost of equity-settled transactions is recognized in statement of comprehensive loss, together with a corresponding increase in equity, during the period which the performance and/or service conditions are to be satisfied, ending on the date on which the relevant employees become fully entitled to the award (the “Vesting Period”). The cumulative expense recognized for equity-settled transactions at the end of each reporting period until the vesting date reflects the extent to which the Vesting Period has expired and the Group's best estimate of the number of equity instruments that will ultimately vest.
If the Company modifies the conditions on which equity-instruments were granted, an additional expense is recognized for any modification that increases the total fair value of the share-based payment arrangement or is otherwise beneficial to the employee/other service provider at the modification date.
| o. | Taxes on income |
As it is not likely that taxable income will be generated in the foreseeable future, deferred tax assets due to accumulated losses is not recognized in the Group's financial statements.
| F-19 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2:- SIGNIFICANT ACCOUNTING POLICIES (Cont.)
| p. | Loss per share |
Losses per share are calculated by dividing the net loss attributable to equity holders of the Company by the weighted number of ordinary shares outstanding during the period. Potential ordinary shares (warrants and unlisted options) are only included in the computation of diluted loss per share when their conversion increases loss per share from continuing operations. Potential ordinary shares that are converted during the period are included in diluted loss per share only until the conversion date and from that date in basic loss per share. The Company's share of loss of subsidiary is included based on the loss per share of OphthaliX multiplied by the number of shares held by the Company.
| q. | Changes in estimates: |
In November 2014, the staff of the Israel Securities Authority issued Accounting Position Paper No. 21-1 regarding the existence in Israel of a deep market in high quality corporate bonds (the "Position Paper") for the purpose of determining, in accordance with IAS 19, the discount rate to be used for defined benefit obligations and other long-term benefits in the Israeli currency. According to the Position Paper, the transition from the use of yields based on Government bonds (2.34%) to market yields based on high quality corporate bonds linked to the Consumer Price Index (3.11%) should be accounted for prospectively as a change in accounting estimate.
The effects of the change in the above mentioned discount rate are immaterial.
| NOTE 3:- | SIGNIFICANT ACCOUNTING JUDGMENTS, ESTIMATES AND ASSUPMTIONS USED IN THE PREPARATION OF THE FINANCIAL STATEMENTS |
In the process of applying the significant accounting policies, the Group has made the following judgments which have the most significant effect on the amounts recognized in the financial statements:
| - | Estimates and assumptions |
The preparation of the financial statements requires management to make estimates and assumptions that have an effect on the application of the accounting policies and on the reported amounts of assets, liabilities and expenses. Changes in accounting estimates are reported in the period of the changes in estimates.
The key assumptions made in the financial statements concerning uncertainties at the end of the reporting period and the critical estimates computed by the Group that may result in a material adjustment to the carrying amounts of assets and liabilities within the next financial year are discussed below.
| - | Determining the fair value of share-based payment transactions |
The fair value of share-based payment transactions is determined using an acceptable option-pricing model. The model includes data as to the share price and exercise price, and assumptions regarding expected volatility, expected life, expected dividend and risk-free interest rate.
| F-20 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| NOTE 3:- | SIGNIFICANT ACCOUNTING JUDGMENTS, ESTIMATES AND ASSUPMTIONS USED IN THE PREPARATION OF THE FINANCIAL STATEMENTS (Cont.) |
| - | Post-employment benefits |
The liability in respect of post-employment defined benefit plans is determined using actuarial valuations. The actuarial valuation involves making assumptions about, among others, discount rates, expected rates of return on assets, future salary increases and mortality rates. The carrying amount of the liability may be significantly affected by changes in such estimates.
| - | Legal claims: |
In estimating the likelihood of outcome of legal claims filed against the Company and its investees, the companies rely on the opinion of their legal counsel. These estimates are based on the legal counsel's best professional judgment, taking into account the stage of proceedings and legal precedents in respect of the different issues. Since the outcome of the claims will be determined in courts, the results could differ from these estimates.
| - | Deferred tax assets: |
Deferred tax assets are recognized for unused carryforward tax losses and deductible temporary differences to the extent that it is probable that taxable profit will be available against which the losses can be utilized. Significant management judgment is required to determine the amount of deferred tax assets that can be recognized, based upon the timing and level of future taxable profits, its source and the tax planning strategy.
NOTE 4:- DISCLOSURE OF NEW STANDARDS IN THE PERIOD PRIOR TO THEIR ADOPTION
| a. | In July 2014, the IASB issued the final and complete version of IFRS 9, "Financial Instruments" ("IFRS 9"), which replaces IAS 39, "Financial Instruments: Recognition and Measurement". IFRS 9 mainly focuses on the classification and measurement of financial assets and it applies to all assets in the scope of IAS 39. |
According to IFRS 9, all financial assets are measured at fair value upon initial recognition. In subsequent periods, debt instruments are measured at amortized cost only if both of the following conditions are met:
| - | the asset is held within a business model whose objective is to hold assets in order to collect the contractual cash flows. |
| - | the contractual terms of the financial asset give rise on specified dates to cash flows that are solely payments of principal and interest on the principal amount outstanding. |
| F-21 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 4:- DISCLOSURE OF NEW STANDARDS IN THE PERIOD PRIOR TO THEIR ADOPTION (Cont.)
Subsequent measurement of all other debt instruments and financial assets should be at fair value. IFRS 9 establishes a distinction between debt instruments to be measured at fair value through profit or loss and debt instruments to be measured at fair value through other comprehensive income.
Financial assets that are equity instruments should be measured in subsequent periods at fair value and the changes recognized in profit or loss or in other comprehensive income (loss), in accordance with the election by the Company on an instrument-by-instrument basis. If equity instruments are held for trading, they should be measured at fair value through profit or loss.
According to IFRS 9, the provisions of IAS 39 will continue to apply to derecognition and to financial liabilities for which the fair value option has not been elected.
Changes in fair values of financial liabilities which are attributable to the change in credit risk should be presented in other comprehensive income. All other changes in fair value should be presented in profit or loss. According to IFRS 9, changes in fair values of financial liabilities which are attributable to the change in credit risk should be presented in other comprehensive income. All other changes in fair value should be presented in profit or loss.
IFRS 9 also prescribes new hedge accounting requirements.
IFRS 9 is to be applied for annual periods beginning on January 1, 2018. Early adoption is permitted. The Company is still evaluating the effect on the above on its financial statements, but it believes at this stage that this effect, if any, is not expected to be material.
| b. | Amendments to IAS 7, "Statement of Cash Flows", regarding additional disclosures of financial liabilities: |
In January 2016, the IASB issued amendments to IAS 7, "Statement of Cash Flows", which require additional disclosures regarding financial liabilities. The amendments require disclosure of the changes between the opening balance and the closing balance of financial liabilities, including changes from cash flows, changes arising from obtaining or losing control of subsidiaries, the effect of changes in foreign exchange rates and changes in fair value.
The amendments are effective for annual periods beginning on or after January 1, 2017. Comparative information for periods prior to the effective date of the amendments is not required. Early application is permitted. The Company will include the necessary disclosures in the financial statements when applicable.
| c. | IFRS 15, "Revenue from Contracts with Customers": |
In May 2014, the IASB issued IFRS 15 ("IFRS 15").
IFRS 15 replaces IAS 18, "Revenue", IAS 11, "Construction Contracts", IFRIC 13, "Customer Loyalty Programs", IFRIC 15, "Agreements for the Construction of Real Estate", IFRIC 18, "Transfers of Assets from Customers" and SIC-31, "Revenue - Barter Transactions Involving Advertising Services".
| F-22 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 4:- DISCLOSURE OF NEW STANDARDS IN THE PERIOD PRIOR TO THEIR ADOPTION (Cont.)
The IFRS 15 introduces a five-step model that will apply to revenue earned from contracts with customers:
Step 1: Identify the contract with a customer, including reference to contract combination and accounting for contract modifications.
Step 2: Identify the separate performance obligations in the contract
Step 3: Determine the transaction price, including reference to variable consideration, financing components that are significant to the contract, non-cash consideration and any consideration payable to the customer.
Step 4: Allocate the transaction price to the separate performance obligations on a relative stand-alone selling price basis using observable information, if it is available, or using estimates and assessments.
Step 5: Recognize revenue when the entity satisfies a performance obligation over time or at a point in time.
IFRS 15 is to be applied retrospectively for annual periods beginning on or after January 1, 2018. Early adoption is permitted. IFRS 15 allows an entity to choose to apply a modified retrospective approach, according to which IFRS 15 will only be applied in the current period presented to existing contracts at the date of initial application. No restatement of comparative periods is required.
The Company is still evaluating the effect on the above on its financial statements, but it believes at this stage that this effect, if any, is not expected to be material.
| d. | IFRS 16, "Leases" |
IFRS 16 replaces International Accounting Standard 17 - Leases (IAS 17) and its related interpretations. The standard's instructions annul the existing requirement from lessees to classify leases as operating or finance leases. Instead of this, for lessees, the new standard presents a unified model for the accounting treatment of all leases according to which the lessee has to recognize an asset and liability in respect of the lease in its financial statements. Similarly, the standard determines new and expanded disclosure requirements from those required at present. The standard will become effective for annual periods as of January 1, 2019, with the possibility of early adoption, so long as the company has also The Company is still evaluating the effect on the above on its financial statements, but it believes at this stage that this effect, if any, is not expected to be material.
| F-23 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 5:- ACCOUNTS RECEIVABLE AND PREPAID EXPENSES
| December 31, | |||||||||
| 2016 | 2015 | ||||||||
| NIS in thousands | |||||||||
| Government authorities | 84 | 242 | |||||||
| Prepaid expenses and others | 7,580 | 2,177 | |||||||
| 7,664 | 2,419 | ||||||||
NOTE 6:- PROPERTY, PLANT AND EQUIPMENT, NET
Balance as of December 31, 2016:
| Laboratory equipment | Computers, office furniture and equipment | Leasehold improvements | Total | ||||||||||||||
| NIS in thousands | |||||||||||||||||
| Cost: | |||||||||||||||||
| Balance at January 1, 2016 | 974 | 1,162 | 646 | 2,782 | |||||||||||||
| Purchases during the year | 3 | 37 | - | 40 | |||||||||||||
| Sale of fixed assets | - | (3 | ) | - | (3 | ) | |||||||||||
| Balance at December 31, 2016 | 977 | 1,196 | 646 | 2,819 | |||||||||||||
| Accumulated depreciation: | |||||||||||||||||
| Balance at January 1, 2016 | 867 | 1,042 | 637 | 2,546 | |||||||||||||
| Depreciation during the year | 13 | 56 | 2 | 71 | |||||||||||||
| Sale of fixed assets | - | (3 | ) | - | (3 | ) | |||||||||||
| Balance at December 31, 2016 | 880 | 1,095 | 639 | 2,614 | |||||||||||||
| Depreciated cost at December 31, 2016 | 97 | 101 | 7 | 205 | |||||||||||||
Balance as of December 31, 2015:
| Laboratory equipment | Computers, office furniture and equipment | Leasehold improvements | Total | ||||||||||||||
| NIS in thousands | |||||||||||||||||
| Cost: | |||||||||||||||||
| Balance at January 1, 2015 | 883 | 1,086 | 646 | 2,615 | |||||||||||||
| Purchases during the year | 91 | 86 | - | 177 | |||||||||||||
| Sale of fixed assets | - | (10 | ) | - | (10 | ) | |||||||||||
| Balance at December 31, 2015 | 974 | 1,162 | 646 | 2,782 | |||||||||||||
| Accumulated depreciation: | |||||||||||||||||
| Balance at January 1, 2015 | 851 | 997 | 634 | 2,482 | |||||||||||||
| Depreciation during the year | 16 | 45 | 3 | 64 | |||||||||||||
| Balance at December 31, 2015 | 867 | 1,042 | 637 | 2,546 | |||||||||||||
| Depreciated cost at December 31, 2015 | 107 | 120 | 9 | 236 | |||||||||||||
| F-24 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 7:- OTHER ACCOUNTS PAYABLE
| December 31, | |||||||||
| 2016 | 2015 | ||||||||
| NIS in thousands | |||||||||
| Employees and payroll accruals | 401 | 583 | |||||||
| Accrued expenses | 3,187 | 3,696 | |||||||
| 3,588 | 4,279 | ||||||||
NOTE 8:- FINANCIAL INSTRUMENTS
| a. | Classification of financial assets and liabilities |
The financial assets and financial liabilities in the statement of financial position are classified by groups of financial instruments pursuant to IAS 39:
| December 31, | |||||||||
| 2016 | 2015 | ||||||||
| NIS in thousands | |||||||||
| Financial assets: | |||||||||
| Accounts receivable and prepaid expenses | 84 | 242 | |||||||
| Financial liabilities: | |||||||||
| Trade payable | 4,804 | 1,803 | |||||||
| Other account payable | 3,588 | 4,279 | |||||||
| Deferred revenues | 5,747 | 4,498 | |||||||
| Warrants exercisable into shares | 10,068 | 16,725 | |||||||
| 24,207 | 27,305 | ||||||||
| b. | Financial risks factors |
The Group's activities expose it to foreign exchange risk. The Group's comprehensive risk management plan focuses on activities that reduce to a minimum any possible adverse effects on the Group's financial performance.
The Company's management identifies and manages financial risks.
| c. | Foreign exchange risk |
The Group is exposed to foreign exchange risk resulting from the exposure to different currencies, mainly the U.S. dollar. Foreign exchange risk arises on recognized assets and liabilities that are denominated in a foreign currency other than the functional currency.
The Group acts to reduce the foreign exchange risk by managing an adequate part of the available liquid sources in or linked to the dollar.
| d. | Fair value |
The carrying amount of cash and cash equivalents, accounts receivable, trade payables and other accounts payable approximate their fair value.
| F-25 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 8:- FINANCIAL INSTRUMENTS (Cont.)
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. To increase the comparability of fair value measures, the following hierarchy prioritizes the inputs to valuation methodologies used to measure fair value:
| Level 1 - | Valuations based on unadjusted quoted prices for identical assets and liabilities in active markets. |
| Level 2 - | Valuations based on observable inputs other than quoted prices included in Level 1, such as quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar assets and liabilities in markets that are not active, or other inputs that are observable or can be corroborated by observable market data. |
| Level 3 - | Valuations based on unobservable inputs reflecting assumptions, consistent with reasonably available assumptions made by other market participants. These valuations require significant judgment. |
The Company's warrants exercisable into shares liability are classified as level 3 in the fair value hierarchy, and measured at fair value on a recurring basis.
Fair value measurements using significant unobservable inputs (Level 3):
| NIS in thousands | |||||
| Balance at December 31, 2014 | 6,969 | ||||
| Fair value of warrants at issuance date | 16,669 | ||||
| Changes in values of warrants exercisable into shares liability | (6,913 | ) | |||
| Balance at December 31, 2015 | 16,725 | ||||
| Fair value of warrants at issuance date | |||||
| Changes in values of warrants exercisable into shares liability | (6,657 | ) | |||
| Balance at December 31, 2016 | 10,068 | ||||
The fair value of warrants granted was valued by using the Black-Scholes call option pricing model. Fair values were estimated using the following assumptions for the warrants call option (range of annualized percentages):
| December 31, | |||||||||
| 2016 | 2015 | ||||||||
| Dividend yield | 0 | 0 | |||||||
| Expected volatility | 59.03%-91.41% | 78.98%-103.85% | |||||||
| Risk-free interest | 0.9%-1.77% | 1.1%-1.81% | |||||||
| Expected life | 1.19-4.28 | 2.19-5.29 | |||||||
| F-26 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 8:- FINANCIAL INSTRUMENTS (Cont.)
| e. | Linkage terms of financial instruments |
| December 31, 2016 | |||||||||||||||||||||
In or linked to dollar | In or linked to Euro | In or linked to GBP | Unlinked | Total | |||||||||||||||||
| NIS in thousands | |||||||||||||||||||||
| Assets: | |||||||||||||||||||||
| Cash and cash equivalents | 30,630 | 100 | - | 473 | 31,203 | ||||||||||||||||
| Accounts receivable | - | - | - | 84 | 84 | ||||||||||||||||
| 30,630 | 100 | - | 557 | 31,287 | |||||||||||||||||
| Liabilities: | |||||||||||||||||||||
| Trade payables | 4,422 | - | 169 | 213 | 4,804 | ||||||||||||||||
| Other accounts payable | 1,870 | 82 | - | 1,636 | 3,588 | ||||||||||||||||
| Warrants exercisable into shares | 10,068 | - | - | - | 10,068 | ||||||||||||||||
| 16,360 | 82 | (169 | ) | 1,849 | 18,460 | ||||||||||||||||
| Financial instruments, net | 14,270 | 18 | (169 | ) | (1,292 | ) | 12,827 | ||||||||||||||
| December 31, 2015 | |||||||||||||||||
In or linked to dollar | In or linked to Euro | Unlinked | Total | ||||||||||||||
| NIS in thousands | |||||||||||||||||
| Assets: | |||||||||||||||||
| Cash and cash equivalents | 64,483 | 139 | 1,404 | 66,026 | |||||||||||||
| Accounts receivable and prepaid expenses | - | - | 242 | 242 | |||||||||||||
| 64,483 | 139 | 1,646 | 66,268 | ||||||||||||||
| Liabilities: | |||||||||||||||||
| Trade payables | 1,418 | 204 | 181 | 1,803 | |||||||||||||
| Other accounts payable | 2,317 | 231 | 1,731 | 4,279 | |||||||||||||
| Warrants exercisable into shares | 16,725 | - | - | 16,725 | |||||||||||||
| 20,460 | 435 | 1,912 | 22,807 | ||||||||||||||
| Financial instruments, net | 44,023 | (296 | ) | (266 | ) | 43,461 | |||||||||||
| F-27 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 8:- FINANCIAL INSTRUMENTS (Cont.)
| f. | Sensitivity tests relating to changes in market factors |
| December 31, | |||||||||
| 2016 | 2015 | ||||||||
| NIS in thousands | |||||||||
| Sensitivity test to changes in the U.S. dollar exchange rate: | |||||||||
| Gain (loss) from the change on financial instruments: | |||||||||
| Increase of 10% in exchange rate | 1,427 | 4,402 | |||||||
| Decrease of 10% in exchange rate | (1,427 | ) | (4,402 | ) | |||||
| Sensitivity test to changes in the market price of listed securities: | |||||||||
| Gain (loss) from the change: | |||||||||
| Increase of 10% in market price | (1,539 | ) | (2,368 | ) | |||||
| Decrease of 10% in market price | 1,472 | 2,304 | |||||||
| * | According to binomial model 10% increase in the market price of listed securities will increase the price of warrants exercisable into shares by approximately 15% and 10% decrease in the market price of listed securities will decrease the price of warrants exercisable into shares by approximately 15%. |
Sensitivity tests and the main work assumptions:
The selected changes in the relevant risk variables were determined based on management's estimate as to reasonable possible changes in these risk variables.
The Group has performed sensitivity tests of principal market risk factors that are liable to affect its reported operating results or financial position. The sensitivity tests present the statement of comprehensive loss in respect of each financial instrument for the relevant risk variable chosen for that instrument as of each reporting date. The test of risk factors was determined based on the materiality of the exposure of the operating results or financial condition of each risk with reference to the functional currency and assuming that all the other variables are constant.
Based on the Group's policy, the Group generally mitigates the currency risk arising from recognized assets and recognized liabilities denominated in foreign currency other than the functional currency by maintaining part of the available liquid sources in deposits in foreign currency. Accordingly, the main currency exposures presented in the sensitivity tables are for those deposits.
| F-28 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 9:- EMPLOYEE BENEFIT LIABILITIES, NET
Employee benefits consist post-employment benefits.
According to the labor laws and Severance Pay Law in Israel, the Company is required to pay compensation to an employee upon dismissal or retirement or to make current contributions in defined contribution plans pursuant to section 14 to the Severance Pay Law, as specified below. The Company's liability is accounted for as a post-employment benefit. The computation of the Company's employee benefit liability is made in accordance with a valid employment contract based on the employee's salary and employment term which establish the entitlement to receive the compensation.
In 2009, management accepted a decision according to which although section 14 applies, as above, the Company would pay all compensation upon dismissal of employees pursuant to the conditions of the Severance Pay Law.
In accordance with the above, since 2009, the Group does not contribute to defined contribution plans, but only to defined benefit plans.
In 2016, the Company's management changed its accounting policy and revoked the previous decision made in 2009 when employee benefit liability was recognized. Accordingly, the cumulative effect of NIS 630 thousand was adjusted and recorded in the statements of comprehensive loss.
NOTE 10:- CONTINGENT LIABILITIES AND COMMITMENTS
| a. | Liabilities to pay royalties: |
| 1. | According to the license agreement that the Company entered into with the NIH on January 29, 2003, the Company was committed to pay royalties until the expiration of the last patent licensed under the license agreement. The last patent under this agreement expired on June 29, 2015, and therefore except with respect to any amounts already accrued on the Company’s balance sheet, no future payments or royalties will be due. |
As of December 31, 2016, the Company accrued NIS 961 thousand (approximately $250 thousand) in Other accounts payable with respect to the NIH.
| 2. | According to the patent license agreement that the Company entered into with Leiden University in the Netherlands on November 2, 2009, which is affiliated with the NIH, the Company was granted an exclusive license for the use of the patents of several compounds, including CF602 in certain territories. |
As of December 31 2016, no accrual is recorded with respect to Leiden University.
| b. | Commitments and license agreements: |
| 1. | On September 22, 2006, the Company signed an exclusive license agreement regarding inflammatory indicators, including rheumatoid arthritis indicators (excluding eye disease indicators) with a public Japanese company, Seikagaku Corporation (the “Japanese Corporation”), for the use, development and marketing of the Company's CF101 drug in Japan only. Under the agreement, the Company received certain payments from the Japanese Corporation. |
| F-29 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 10:- CONTINGENT LIABILITIES AND COMMITMENTS (Cont.)
In August 2015, the Company and the Japanese Corporation entered into an agreement terminating the license agreement.
| 2. | In March 2015, the Company signed a distribution agreement with Cipher. As part of the distribution agreement, Cipher will distribute Can-Fite's lead drug candidate, CF101 ("Product") for the treatment of psoriasis and rheumatoid arthritis in the Canadian market upon receipt of regulatory approvals. |
Under the terms of the agreement, Cipher made an upfront payment of NIS 5,141 thousand (CAD 1,650 thousand) to the Company in March 2015. In addition, the agreement provides that additional payments of up to CAD 2,000 thousand will be received by the Company upon the achievement of certain milestones plus royalty payments of 16.5% of net sales of CF101 in Canada. The agreement further provides that the Company will deliver finished Product to Cipher and that Cipher will reimburse the Company for the cost of manufacturing.
Furthermore, under the distribution agreement, the Company shall be responsible for conducting Product development activities including management of the clinical studies required in order to secure regulatory approvals, and shall use commercially reasonable efforts in conducting such activities. In addition the Company obliged to form a joint steering committee with Cipher which will oversee the progress of the clinical studies.
The Company identified four components in the agreement: (i) performing the research and development services through regulatory approval; (ii) exclusive license to distribute the product in Canada; (iii) participation in joint steering committee; and, (iv) royalties resulting from future sales of the product. Components (i) – (iii) were analyzed as one unit of accounting. Consequently, revenue from these components is recorded based on the term of the research and development services (which is the last deliverable in the arrangement). The Company estimates these services will spread over a period of 24 quarters beginning March 2015. Component (iv) was not accounted as part of the research and development services and will be recognized entirely upon the Company reaching sales stage.
| 3. | In October 2016, the Company signed a distribution agreement with Chong Kun Dang Pharmaceuticals Corp. ("CKD") for future sales in South Korea. As part of the distribution agreement, CKD will distribute the company's lead drug candidate, CF102 ("Product") for the treatment of liver cancer in the South Korean market upon receipt of regulatory approvals. |
Under the terms of the agreement, CKD made an upfront payment of NIS 1,901 thousand ($ 500 thousand) to the Company in December 2016. In addition, the agreement provides that additional payments of up to $ 2,500 thousand will be received by the Company upon the achievement of certain milestones plus royalty payments of 23% of net sales of CF102 in South Korea. The agreement further provides that the Company will deliver finished Product to CKD and that CKD will reimburse the Company for the cost of manufacturing. The Company identified four components in the agreement: (i) performing the research and development services through regulatory approval; (ii) exclusive license to distribute the product in South Korea;
| F-30 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 10:- CONTINGENT LIABILITIES AND COMMITMENTS (Cont.)
(iii) participation in joint steering committee; and, (iv) royalties resulting from future sales of the product. Components (i) – (iii) were analyzed as one unit of accounting. Consequently, revenue from these components is recorded based on the term of the research and development services (which is the last deliverable in the arrangement). The Company estimates these services will spread over a period of 20 quarters beginning October 2016. Component (iv) was not accounted as part of the research and development services and will be recognized entirely upon the Company reaching sales stage.
| 4. | On December 22, 2008, the Company signed an agreement regarding the provision of a license for its CF101 drug with a South Korean pharmaceutical company, Kwang Dong Pharmaceutical Co. Ltd. (the “Korean License Agreement” and the “Korean Company”, respectively). According to the license agreement, the Company granted the Korean Company a license to use, develop and market its CF101 drug for treating only rheumatoid arthritis only in the Republic of Korea. |
As of December 31, 2016, the Company estimates that such contingent payments are remote.
| 5. | Lease commitments: |
The Company lease motor vehicles through operating leases. The lease is for a period ending September 2019. Future minimum lease commitments under non-cancelable operating leases as of December 31, 2016 are as follows:
| NIS in thousands | |||||
| 2017 | 154 | ||||
| 2018 | 114 | ||||
| 2019 | 36 | ||||
| 304 | |||||
Lease expenses for the years ended December 31, 2014, 2015 and 2016 were approximately NIS 185 thousand, NIS 169 thousand and NIS 195 thousand, respectively.
| c. | Class action: |
On June 29, 2015 the Company was served with a motion to approve a purported class action, naming the Company, its Chief Executive Officer and its directors as defendants. The motion was filed with the District Court of Tel-Aviv. The lawsuit alleges, among other things, that the Company misled the public with regard to disclosures concerning the efficacy of the Company’s drug candidate, CF101.
The claimant alleges that he suffered personal damages of over NIS 73 thousand, while also claiming that the shareholders of the Company suffered damages of approximately NIS 125 million. The Company believes it has strong defense against these allegations and that the District Court should deny the motion to approve the class action, however, there is no assurance that the Company’s position will be accepted by the District Court. In such case the Company may have to divert attention of its executives to deal with this class action as well as incur expenses that may be beyond its insurance coverage for such cases, which cause a risk of loss and expenditures that may adversely affect its financial condition and results of operations.
| F-31 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 11:- EQUITY
| a. | Composition of share capital: |
| December 31, 2016 | December 31, 2015 | ||||||||||||||||
| Authorized | Issued and outstanding | Authorized | Issued and outstanding | ||||||||||||||
| Number of Shares | |||||||||||||||||
| Ordinary shares of NIS 0.25 par value each | 80,000,000 | 28,156,728 | 80,000,000 | 28,119,728 | |||||||||||||
| b. | On December 3, 2015, a Special General Meeting of Shareholders of Can-Fite BioPharma Ltd. (the "Company") approved, in accordance with the majority required, a proposal to increase the Company's authorized share capital by NIS 10,000,000 such that following the increase, the authorized share capital shall equal NIS 20,000,000 divided into 80,000,000 ordinary shares, par value NIS 0.25 each, and to amend the Company's articles of association accordingly. |
| c. | Issued and outstanding capital: |
| Number of shares | NIS par value | ||||||||
| Balance at December 31, 2014 | 21,763,404 | 5,440,851 | |||||||
| Issuance of share capital | 6,356,324 | 1,589,081 | |||||||
| Balance at December 31, 2015 | 28,119,728 | 7,029,932 | |||||||
| Issuance of share capital | 37,000 | 9,250 | |||||||
| Balance at December 31, 2016 | 28,156,728 | 7,039,182 | |||||||
| d. | Ordinary shares and rights attached to shares: |
On May 2, 2013, the annual general meeting of the Company's shareholders approved a reverse stock split of one share for each twenty five shares outstanding (1:25) (the “Reverse Split”). The Reverse Split became effective as of the close of business on May 10, 2013. The Company's authorized share capital after the Reverse Split was NIS 10 million divided into 40 million ordinary shares, NIS 0.25 par value per share, of the Company. All ordinary shares, warrants, options, per share data and exercise prices included in these financial statements and notes for all periods presented have been retroactively adjusted to reflect the Reverse Split with respect to the Company’s share capital.
All ordinary shares have equal rights for all intent and purposes and each ordinary share confers its holder:
| 1. | The right to be invited and participate in all the Company's general meetings, both annual and regular, and the right to one vote per ordinary share owned in all votes and in all Company's general meeting participated. |
| F-32 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 11:- EQUITY (Cont.)
| 2 | The right to receive dividends if and when declared and the right to receive bonus shares if and when distributed. |
| 3. | The right to participate in the distribution of the Company's assets upon liquidation. |
| e. | Issue of shares and warrants and changes in equity: |
| 1. | In March 2014, the Company completed a private placement pursuant to which it sold an aggregate of 982,344 ADSs representing 1,964,688 ordinary shares and warrants to purchase an additional 491,172 ADSs representing 982,344 ordinary shares for an aggregate purchase price of NIS 17,567 thousand (the "March 2014 Financing"). For further information regarding the warrants, please refer to Note 11.f.2. |
| 2. | In December 2014, the Company completed a registered direct offering pursuant to which it sold an aggregate of 1,797,753 ADSs representing 3,595,506 ordinary shares. In addition, the Company issued unregistered warrants to purchase 898,877 ADSs representing 1,797,753 ordinary shares. The offering (the "December 2014 Financing") resulted in gross proceeds of NIS 31,923 thousand. For further information regarding the warrants, please refer to Note 11.f.2. |
As part of the March 2014 Financing, the Company also issued placement agent warrants to purchase 49,117 ADSs representing 98,234 ordinary shares exercisable at $ 6.43 per ADS (equivalent to $ 3.215 per ordinary share), subject to certain adjustments, for a period of four years.. In addition, as part of the December 2014 Financing, the Company also issued placement agent warrants to purchase 89,888 ADSs representing 179,775 ordinary shares exercisable at $ 4.45 per ADS (equivalent to $ 2.225 per ordinary share), subject to certain adjustments, for a period of five years. The placement agent warrants issued in the March 2014 Financing may be exercised on a cashless basis and contain registration rights covering the resale of the ordinary shares represented by ADSs underlying the placement agent warrants and the placement agent warrants issued in the December 2014 Financing may be exercised on a cashless basis if six months after issuance there is no effective registration statement registering the ADSs underlying the warrants. The fair value of the placement agents warrants issued in the March 2014 Financing and December 2014 Financing at the grant date were NIS 381 thousand and NIS 613 thousand, respectively, and were considered as additional issuance costs.
The cash issuance costs in relation to the March 2014 Financing and December, 2014 Financing were NIS 1,795 thousand and NIS 3,020 thousand, respectively.
In relation to the issuance of March 2014 Financing and December 2014 Financing, the Company first allocated the proceeds to the warrant, that due to the dollar exercise price terms and in accordance with IAS 39 is being considered a freestanding liability instrument that is measured at fair value at each reporting date, based on its fair value, with changes in the fair values being recognized in the Company's statement of comprehensive loss as financial income or expense. The remaining proceeds were allocated to the shares and were recorded to equity. The issuance costs were allocated between the warrants and the shares in proportion to the allocation of the proceeds. The portions of the issuance costs that were allocated to the warrants and to the ordinary share were recorded as financial expense in the Company's statement of comprehensive loss and to the additional paid in capital in the Company's balance sheet, respectively.
| F-33 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 11:- EQUITY (Cont.)
| 3. | In September 2015, the Company completed a registered direct offering pursuant to which it sold an aggregate 2,068,966 ADSs representing 4,137,932 ordinary shares. In addition, the Company issued unregistered warrants to purchase 1,034,483 ADSs representing 2,068,966 ordinary shares. The offering (the “September 2015 Financing”) resulted in gross proceeds of NIS 34,767 thousand. For further information regarding the warrants, please refer to Note 11.f.3. |
In October 2015, the Company completed a registered direct offering pursuant to which it sold an aggregate 1,109,196 ADSs representing 2,218,392 ordinary shares. In addition, the Company issued unregistered warrants to purchase 443,678 ADSs representing 887,356 ordinary shares. The offering (the “October 2015 Financing”) resulted in gross proceeds of NIS 18,653 thousand. For further information regarding the warrants, please refer to Note 11.f.3.
As part of the September 2015 Financing, the Company also issued placement agent warrants to purchase 103,448 ADSs representing 206,897 ordinary shares exercisable at $ 5.25 per ADS (equivalent to $ 2.625 per ordinary share), subject to certain adjustments, for a period of five years. In addition, as part of the October 2015 Financing, the Company also issued placement agent warrants to purchase 55,460 ADSs representing 110,920 ordinary shares exercisable at $ 5.25 per ADS (equivalent to $ 2.625 per ordinary share), subject to certain adjustments, for a period of five years. The placement agent warrants may be exercised on a cashless basis if six months after issuance there is no effective registration statement registering the ADSs underlying the warrants. The fair value of the placement agents warrants issued in the September 2015 Financing and October 2015 Financing at the grant date were NIS 1,224 thousand and NIS 554 thousand, respectively and were considered as additional issuance costs.
The cash issuance costs in relation to the September 2015 Financing and October 31, 2015 Financing were NIS 3,060 thousand and NIS 2,028 thousand, respectively.
In relation to the September 2015 Financing and October 2015 Financing, the Company first allocated the proceeds to the warrants, that due to the dollar exercise price terms and in accordance with IAS 39 is being considered a freestanding liability instrument that is measured at fair value at each reporting date, based on its fair value, with changes in the fair values being recognized in the Company's statement of comprehensive loss as financial income or expense. The remaining proceeds were allocated to the shares and were recorded to equity. The issuance costs were allocated between the warrants and the shares in proportion to the allocation of the proceeds. The portions of the issuance costs that were allocated to the warrants and to the ordinary share were recorded as financial expense in the Company's statement of comprehensive loss and to the additional paid in capital in the Company's balance sheet, respectively.
| F-34 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 11:- EQUITY (Cont.)
| f. | Warrants classified as liability: |
| 1. | On March 31, 2014, 9,907,500 registered warrants (Series 7) that were exercisable into 396,300 ordinary shares of the Company were expired. Accordingly, the Company recorded an amount of NIS 119 thousand as financial income in its statement of comprehensive loss. |
| 2. | As mentioned in Note 11.e.1 the Company issued warrants as part of the March 2014 Financing and December 2014 Financing. The warrants issued in the March 2014 Financing may be exercised after 6 months from the date of issuance for a period of four years and have an exercise price of $ 6.43 per ADS (equivalent to $ 3.215 per ordinary share) (subject to certain adjustments). The warrants issued in the December 2014 Financing may be exercised for a period of five years following issuance and have an exercise price of $ 4.45 per ADS (equivalent to $ 2.225 per ordinary share) (subject to certain adjustments). The fair value of the warrants issued as part of the March 2014 Financing at the commitment date and December 31, 2015 were NIS 6,127 thousand and NIS 5,315 thousand, respectively. The fair value of the warrants issued as part of the December 2014 Financing at the commitment date and December 31, 2015 were NIS 12,235 thousand and NIS 6,370 thousand, respectively with changes in recorded as financial income in the Company's statement of comprehensive loss. |
| 3. | As mentioned in Note 11.e.2 the Company issued warrants as part of the September 2015 Financing and October 2015 Financing. These warrants may be exercised after 6 months from the date of issuance for a period of five and a half years and have an exercise price of $ 5.25 per ADS (equivalent to $ 2.625 per ordinary share) (subject to certain adjustments). The fair value of the warrants issued as part of the September 2015 Financing at the commitment date and December 31, 2015 were NIS 12,235 thousand and NIS 6,370 thousand, respectively. The fair value of the warrants issued as part of the October 2015 Financing at the commitment date and December 31, 2015 were NIS 4,434 thousand and 2,746 thousand, respectively. Changes in fair value of the warrants from commitment date to December 31, 2015 were recorded as financial income in the Company's statement of comprehensive loss. |
| F-35 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 11:- EQUITY (Cont.)
| g. | Warrants classified as equity: |
The Company had 12,168,000 registered warrants (Series 9) that were exercisable into 486,720 ordinary shares of the Company for the exercise price of NIS 21.25 per share. These warrants expired on May 1, 2015.
The Company has 39,042,000 registered warrants (Series 10) that are exercisable into 1,561,680 ordinary shares of the Company for NIS 9.85 per share. The warrants are exercisable, according to the court approval, until October 31, 2017.
The Company has 37,372,500 registered warrants (Series 11) that are exercisable into 1,494,900 ordinary shares of the Company for NIS 9.80 per share. The warrants are exercisable, according to the court approval, until October 31, 2017.
The Company has 1,470,000 registered warrants (Series 12) that are exercisable into 1,470,000 ordinary shares of the Company for NIS 15.29 per share. The warrants are exercisable, according to the court approval, until October 31, 2017.
As described at Note 11.e.2, in September and October 2015 the company issued warrants to purchase 2,275,863 and 998,276 of the Company's ordinary shares, respectively.
| h. | Unlisted share options: |
On November 28, 2013, the Board of Directors approved the adoption of the 2013 ESOP (the "2013 Plan"). Under the 2013 Plan, the Company may grant its officers, directors, employees and consultants, Stock options, of the Company. Each Stock option granted shall be exercisable at such times and terms and conditions as the Board of Directors may specify in the applicable option agreement, provided that no option will be granted with a term in excess of 10 years.
Upon the adoption of the 2013 ESOP the Company reserved for issuance 1,000,000 shares of Common Stock, NIS 0.25 par value each.
On May 28, 2014 the company’s board of directors approved the extension by one year of options which are exercisable into 502,025 ordinary shares of the company, originally granted to investor on October 2010, till October 21, 2015. As of May 28, 2014, the modification date, the Company has evaluated the incremental fair value to be NIS 331 thousand which was recorded as immediately expense.
| i. | Treasury shares: |
As of December 31, 2016, the Company's shares held by OphthaliX amounted to 446,827 ordinary shares.
| December 31, | |||||||||
| 2016 | 2015 | ||||||||
| % | |||||||||
| Percentage of issued capital | 1.59 | 1.59 | |||||||
| F-36 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 12:- SHARE-BASED PAYMENT TRANSACTIONS
| a. | Expenses recognized in the financial statements: |
| Year ended December 31, | |||||||||||||
| 2016 | 2015 | 2014 | |||||||||||
| NIS in thousand | |||||||||||||
| Research and development expenses | 495 | 207 | 27 | ||||||||||
| General and administrative expenses | 676 | 235 | 799 | ||||||||||
| 1,171 | 442 | 826 | |||||||||||
| b. | Share-based payment transactions granted by the Company: |
| 1. | On March 19, 2015, the Company's board of directors approved a grant of unlisted options exercisable into 40,000 of the Company's ordinary shares to three of its employees and one senior officer for an exercise price of NIS 8.118 per shares. The options will vest on a quarterly basis for a period of 4 years from the grant date. |
| 2. | In October 2015, the Company granted an amount of 200,000 options to acquire up to 200,000 of the Company's ordinary shares to one of its directors at an exercise price of NIS 3.573 per share. The options will vest over a period of three years on a quarterly basis for 12 consecutive quarters from the date of the grant. The term of the options is 10 years. |
| 3. | In February 2016, the Company's board of directors approved a grant of unlisted options exercisable into 160,000 of the Company's ordinary shares to three of its employees and one senior officer for an exercise price of NIS 4.317 per shares. The options vest on quarterly basis for a period of 4 years from the grant date. |
| 4. | In May 2016, the Company's board of directors approved a grant of 74,000 shares of the Company to its service provider. Pursuant to the agreement with the service provider, and as partial consideration, the Company issued 37,000 ordinary shares and agreed to issue an additional 37,000 ordinary shares within 180 days, provided that the agreement was not terminated. As of December 31, 2016 the Company recorded an amount of NIS 322 for share based payment expenses relating to this transaction. |
| 5. | On May 26, 2016 the Company's board of directors approved a grant of 20,000 options exercisable up to 20,000 Ordinary Shares of the Company to one of its advisers at an exercise price of 5.376 NIS per Share. The options will vest on a quarterly basis for a period of 4 years from the grant date. |
| F-37 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 12:- SHARE-BASED PAYMENT TRANSACTIONS (Cont.)
| c. | Movement during the year: |
The following table lists the number of share options, their weighted average exercise prices and modification in option plans of employees, directors and consultants for the periods indicated:
| Shares subject to options outstanding | |||||||||||||||||||||||||
| 2016 | 2015 | 2014 | |||||||||||||||||||||||
| Number | Weighted average exercise price | Number | Weighted average exercise price | Number | Weighted average exercise price | ||||||||||||||||||||
| NIS | NIS | NIS | |||||||||||||||||||||||
| Outstanding at beginning of year | 798,579 | 12.21 | 592,707 | 15.33 | 623,279 | 15.23 | |||||||||||||||||||
| Grants | 180,000 | 4.43 | 240,000 | 4.33 | 10,000 | 12.00 | |||||||||||||||||||
| Exercised | - | - | - | - | (1,656 | ) | 0.25 | ||||||||||||||||||
| Forfeited/expired | (241,551 | ) | 12.79 | (34,128 | ) | 10.96 | (38,916 | ) | 13.61 | ||||||||||||||||
| Outstanding at end of year | 737,028 | 10.12 | 798,579 | 12.21 | 592,707 | 15.33 | |||||||||||||||||||
| Exercisable at end of year | 402,500 | 14.90 | 557,749 | 15.56 | 565,694 | 15.58 | |||||||||||||||||||
| d. | The weighted average remaining contractual life for the shares subject to options outstanding as of December 31, 2016, 2015 and 2014 was 5.86 years, 4.67 years and 3.11 years, respectively. |
| e. | The range of exercise prices for shares subject to options outstanding as of December 31, 2016, 2015 and 2014 was between NIS 0.25 and NIS 31.175. |
| f. | The fair value of the Company's share options granted for the years ended December 31, 2015 and 2016 was estimated using the binomial option pricing model using the following assumptions: |
| December 31, | |||||||||
| Description | 2016 | 2015 | |||||||
| Risk-free interest rate | 2.01%-2.02% | 1.94%-2.17% | |||||||
| Expected volatility | 77.84%-78.22% | 67.97%-77.03% | |||||||
| Dividend yield | 0 | 0 | |||||||
| Contractual life | 10 | 10 | |||||||
| Early Exercise Multiple (Suboptimal Factor) | 2.5 | 2-2.5 | |||||||
| Exercise price | 4.317-5.376 | 3.573-8.1205 | |||||||
| F-38 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 13:- RESEARCH AND DEVELOPMENT EXPENSES
| Year ended December 31, | |||||||||||||
| 2016 | 2015 | 2014 | |||||||||||
| NIS in thousands | |||||||||||||
| Clinical and preclinical trials | 19,504 | 10,815 | 12,295 | ||||||||||
| Salary and related expenses | 2,546 | 2,575 | 1,931 | ||||||||||
| Patents | 759 | 842 | 760 | ||||||||||
| Royalties | 42 | 240 | 510 | ||||||||||
| Laboratory materials | 144 | 190 | 205 | ||||||||||
| Rent | 181 | 182 | 184 | ||||||||||
| Depreciation | 15 | 19 | 9 | ||||||||||
| Others | 189 | 189 | 306 | ||||||||||
| 23,380 | 15,052 | 16,200 | |||||||||||
NOTE 14:- GENERAL AND ADMINISTRATIVE EXPENSES
| Year ended December 31, | |||||||||||||
| 2016 | 2015 | 2014 | |||||||||||
| NIS in thousands | |||||||||||||
| Professional services | 3,917 | 4,408 | 3,420 | ||||||||||
| Investors and public relations | 1,959 | 1,633 | 3,069 | ||||||||||
| Salary and related expenses | 1,590 | 1,669 | 2,089 | ||||||||||
| Directors' fee | 805 | 773 | 691 | ||||||||||
| Rent | 123 | 123 | 123 | ||||||||||
| Travel | 753 | 814 | 850 | ||||||||||
| Insurance | 544 | 469 | 465 | ||||||||||
| Stock exchange fees | 222 | 222 | 317 | ||||||||||
| Office and computer maintenance | 260 | 250 | 258 | ||||||||||
| Vehicle maintenance | 55 | 49 | 89 | ||||||||||
| Depreciation | 56 | 45 | 35 | ||||||||||
| Others | 199 | 178 | 167 | ||||||||||
| 10,483 | 10,633 | 11,573 | |||||||||||
NOTE 15:- FINANCE EXPENSES (INCOME)
| Year ended December 31, | |||||||||||||
| 2016 | 2015 | 2014 | |||||||||||
| NIS in thousands | |||||||||||||
| Finance expenses: | |||||||||||||
| Bank commissions | 105 | 76 | 58 | ||||||||||
| Issuance expenses related to warrants exercisable into shares | - | 2,116 | 1,170 | ||||||||||
| Net loss from exchange rate fluctuations | 580 | - | - | ||||||||||
| Financial expenses from defined benefit plans | - | 11 | - | ||||||||||
| 685 | 2,203 | 1,228 | |||||||||||
| Finance income: | |||||||||||||
| Interest income on bank deposits | (342 | ) | (87 | ) | (45 | ) | |||||||
| Net gain from exchange rate fluctuations | - | (492 | ) | (1,366 | ) | ||||||||
| Net change in fair value warrants exercisable into shares | (6,657 | ) | (6,913 | ) | (3,089 | ) | |||||||
| (6,999 | ) | (7,492 | ) | (4,500 | ) | ||||||||
| F-39 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 16:- LOSS PER SHARE
| a. | Details of the number of shares and loss used in the computation of loss per share: |
| Year ended December 31, | |||||||||||||||||||||||||
| 2016 | 2015 | 2014 | |||||||||||||||||||||||
| Weighted number of shares | Loss | Weighted number of shares | Loss | Weighted number of shares | Loss | ||||||||||||||||||||
In thousands | NIS in thousands | In thousands | NIS in thousands | In thousands | NIS in thousands | ||||||||||||||||||||
| Number of shares and loss used in the computation of basic and diluted loss per share | 27,710 | 26,532 | 22,953 | 18,726 | 17,546 | 23,759 | |||||||||||||||||||
| b. | To compute diluted loss per share for the year ended December 31, 2015 and 2016, the total number of 10,747,904 shares subject to outstanding warrants and 798,582 shares subject to outstanding unlisted options have not been taken into account since they have anti-dilutive effect. |
| F-40 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 17:- TAXES ON INCOME
| a. | Corporate tax rates: |
| 1. | Israeli taxation: |
Corporate tax rate in Israel in 2015 and 2016 is 26.5% and 25%.
On January 4, 2016, the Israeli Parliament's Plenum approved by a second and third reading the Bill for Amending the Income Tax Ordinance (No. 217) (Reduction of Corporate Tax Rate), 2015, which consists of the reduction of the corporate tax rate from 26.5% to 25%.
In December 2016, the Israeli Parliament approved the Economic Efficiency Law (Legislative Amendments for Applying the Economic Policy for the 2017 and 2018 Budget Years), 2016 which reduces the corporate income tax rate to 24% (instead of 25%) effective from January 1, 2017 and to 23% effective from January 1, 2018.
The Company estimates that the effect of the change in tax rates will have no impact on the financial statements.
| 2. | Income tax on non-Israeli subsidiary: |
The corporate tax in the U.S. applying to a Company's subsidiary (incorporated in state of Delaware), consists of a progressive corporate tax at a rate of up to 35% plus state tax and local tax at rates depending on the state and the city in which the company's subsidiary manages its business. In the Company's estimation, it is subject to approximately a 40% tax rate.
| b. | Final tax assessments: |
The Company received final tax assessments through 2012.
The related company, OphthaliX and Eye-Fite, has not received final tax assessments since its incorporation.
| c. | Net operating carryforward losses for tax purposes and other temporary differences: |
As of December 31, 2016 the Company and Eye-Fite had carryforward losses amounting to approximately NIS 325,970 thousand and NIS 10,368 thousand.
OphthaliX is subject to U.S. income taxes. As of December 31, 2016, the Company has net operating loss carryforwards for federal income tax purposes of approximately $ 2,385 thousand (approximately NIS 9,170 thousand) which will expire in the years 2019 to 2036. The Company has no operating loss carry forwards for state income tax purposes.
| d. | Deferred taxes |
The Company did not recognize deferred tax assets for carryforward losses and other temporary differences because their utilization in the foreseeable future is not probable.
| F-41 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 18:- TRANSACTIONS WITH RELATED PARTIES
| a. | The related parties of the Company are associates, subsidiaries, directors and key management personnel of the Group, and a close member of the family of any of the persons mentioned above. |
| b. | The Chairman of the Company's board of directors is a senior partner in the patent firm which represents the Company in intellectual property and commercial matters (the "Service Provider"). The Service Provider charges the Company for services it renders on an hourly basis. |
| c. | Composition of balances with related parties as of December 31, 2016 and December 31, 2015: |
| As of December 31, | |||||||||
| 2016 | 2015 | ||||||||
| NIS in thousands | |||||||||
| Patent expenses(1) | 19 | 105 | |||||||
| Directors' fee (2) | 156 | 128 | |||||||
| (1) Number of related parties | 1 | 1 | |||||||
| (2) Number of directors | 4 | 4 | |||||||
| d. | Composition of balances with related parties for the year ended December 31, 2016, and each of the three years then ended: |
| Year ended December 31, | |||||||||||||
| 2016 | 2015 | 2014 | |||||||||||
| NIS in thousands | |||||||||||||
| Management and consulting fees (including bonuses) (1) | 1,238 | 1,128 | 1,312 | ||||||||||
| Other expenses and share-based payment (1) | 608 | 252 | 57 | ||||||||||
| Patent expenses | 759 | 805 | 793 | ||||||||||
| Directors' fee and share-based payment (2) | 514 | 575 | 417 | ||||||||||
| (1) Number of related parties | 1 | 1 | 1 | ||||||||||
| (2) Number of directors | 5 | 5 | 4 | ||||||||||
| e. | Eye-Fite License agreement |
A license agreement was entered into between the Company and Eye-Fite (the “Eye-Fite License Agreement”) according to which the Company granted Eye-Fite a non transferable exclusive license for the use of the Company's know-how solely in the field of ophthalmic diseases for research, development, commercialization and marketing throughout the world.
| F-42 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 18:- TRANSACTIONS WITH RELATED PARTIES (Cont.)
Eye-Fite is permitted to sublicense subject to the Eye-Fite License Agreement. Prior to the transfer of Eye-Fite to OphthaliX described above, as consideration for the grant of the license, the Company received 1,000 ordinary shares of Eye-Fite, par value NIS 0.01 per share, representing 100% of the issued and outstanding share capital of Eye-Fite.
In connection with the Eye-Fite License Agreement, referred to above, Eye-Fite was obligated to make to the U.S. National Institutes of Health ("NIH"), with regard to the patents of which are included in the license to Eye-Fite, for as long as the license agreement between Can-Fite and NIH remains in effect, a nonrefundable minimum annual royalty fee and potential future royalties of 4.0% to 5.5% on net sales. In addition, the Company was obligated to make certain milestone payments to NIH ranging from $ 25 thousand to $ 500 thousand upon the achievement of various development milestones for each indication. In June 2015, the license agreement between Can-Fite and NIH terminated due to patent expiration.
All inventions resulting from the indication that is licensed under the Eye-Fite License Agreement shall belong to the Company whether it was invented solely by it, solely by Eye-Fite or by both in cooperation. However, the Company granted Eye-Fite an exclusive license to use these inventions in the field of ophthalmic diseases around the world at no consideration. The Eye-Fite License Agreement will remain in effect until the expiration of the last patent licensed thereunder unless it is terminated sooner by a mutual agreement in writing or by one of the parties according to the clauses of the Eye-Fite License Agreement.
| f. | Eye-Fite Services Agreement |
In addition to the Eye-Fite License Agreement, the Company, OphthaliX and Eye-Fite entered into a services agreement (the “Services Agreement”) pursuant to which the Company provides management services with respect to all pre-clinical and clinical research studies, production and supply of the compounds related to the Eye-Fite License Agreement and payment for consultants that are listed in the agreement for their involvement in the clinical trials and in all the activities leading up to, and including, the commercialization of CF101 for ophthalmic indications.
As consideration for rendering the services, the Company shall be reimbursed for its costs and expenses incurred in rendering the services plus 15%, as well as reimbursed for the expenses actually charged for the maintenance of patents underlying the license to Eye-Fite.
In addition, in February 2013, as last updated in August 2015, the Company issued to OphthaliX a formal letter stating that the Company agrees to defer payments owing to it under the Services Agreement from January 31, 2013 for the performance of the clinical trials of CF101 in ophthalmic indications until the completion of fundraising by OphthaliX sufficient to cover such deferred payments. Also, in August 2015, the Company issued a financial support letter pursuant to which it committed to cover any shortfall in the costs and expenses of operations of OphthaliX which are in excess of the OphthaliX’s available cash to finance its operations, including cash generated from any future sale of Company shares held by OphthaliX.
| F-43 |
CAN-FITE BIOPHARMA LTD. AND ITS SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 18:- TRANSACTIONS WITH RELATED PARTIES (Cont.)
Both letters expired in October 2016. On November 14, 2016 the Company agreed to extend the support letter under the same terms and conditions in order to fund Ophthalix's operations. Such support letter expired on February 28, 2017. Deferred payments under the Services Agreement are currently due. Any related balance shall bear an interest at a rate of 3% per annum. As of December 31, 2016, the deferred payments to the Company totaled $ 4,459 thousand.
Further, the Company will be entitled to an additional payment of 2.5% of any revenues received by OphthaliX or any of its affiliates for the rights to use the transferred know-how (the “Additional Payment”).
The Company is entitled during a 5-year period from the date of the approval of the Services Agreement, to convert its right to the Additional Payment into 480,023 shares of OphthaliX (representing about 5% of OphthaliX shares on a fully diluted basis as of the date of closing the spin-off agreement) in consideration for the exercise price set forth in the services agreement. The Services Agreement shall remain in force for an unlimited period of time.
NOTE 19:- SUBSEQUENT EVENTS
In January 2017, the Company completed a registered direct offering pursuant to which it sold an aggregate 2,500,000 ADSs representing 5,000,000 ordinary shares. In addition, the Company issued unregistered warrants to purchase 1,250,000 ADSs representing 2,500,000 ordinary shares. The warrants may be exercised after six months from issuance for a period of five and a half years from issuance at an exercise price of $2.25 per ADS, subject to adjustment as set forth therein. The warrants may be exercised on a cashless basis if six months after issuance there is no effective registration statement registering the ADSs underlying the warrants.The offering (the “January 2017 Financing”) resulted in gross proceeds of NIS 19,050 thousand.
As part of the January 2017 Financing, the Company also issued placement agent warrants to purchase 125,000 ADSs representing 250,000 ordinary shares exercisable at $2.25, subject to certain adjustments, for a period of five years. The placement agent warrants may be exercised on a cashless basis if six months after issuance there is no effective registration statement registering the ADSs underlying the warrants.
| F-44 |
Index to Exhibits
| Exhibit No. | Description | |
| 1.1 | Amended and Restated Articles of Association of Can-Fite BioPharma Ltd (1) | |
| 2.1 | Form of Amended and Restated Deposit Agreement, by and among Can-Fite BioPharma Ltd., The Bank of New York Mellon and the Owners and Holders of American Depositary Shares, dated September 11, 2013 (2) | |
| 4.1 | Employment and Non-Competition Agreement with Motti Farbstein, dated June 10, 2003 (3) | |
| 4.2 | Consulting Agreement with BioStrategics Consulting, Ltd, dated September 27, 2005 (3) | |
| 4.3 | Service Management Agreement with F.D. Consulting International and Marketing Ltd., dated June 27, 2002 (3) | |
| 4.4 | Master Services Agreement with Accellient Partners, dated May 10, 2010 (3) | |
| 4.5 | License Agreement, by and between The University of Leiden and Can-Fite BioPharma Ltd., dated November 2, 2009 (3) | |
| 4.6 | License Agreement, by and between Kwang Dong Pharmaceutical Co., Ltd. and Can-Fite BioPharma Ltd., dated December 14, 2008 (3) | |
| 4.7 | License Agreement, by and between Eye-Fite, Ltd. and Can-Fite BioPharma Ltd., dated November 21, 2011 (3) | |
| 4.8 | Services Agreement, by and among Denali Concrete Management Inc., Eye-Fite Ltd. and Can-Fite BioPharma Ltd., dated November 21, 2011 (3) | |
| 4.9 | Letter from Can-Fite BioPharma Ltd. to OphthaliX, Inc. regarding “Reimbursement for the Costs of the Clinical Trial”, dated February 24, 2013 (3) | |
| 4.10 | Agreement, by and between Denali Concrete Management Inc. and Can-Fite BioPharma Ltd., dated November 21, 2011 (3) | |
| 4.11 | Stock Purchase Agreement, by and between Denali Concrete Management Inc. and Can-Fite BioPharma Ltd., dated November 21, 2011 (3) | |
| 4.12 | Subscription Agreement, by and between Denali Concrete Management Inc. and Can-Fite BioPharma Ltd., dated November 21, 2011 (3) | |
| 4.13 | Common Stock Purchase Warrant issued by Denali Concrete Management Inc. to Can-Fite BioPharma Ltd., dated November 21, 2011 (3) | |
| 4.14 | Can-Fite BioPharma Ltd. 2003 Israeli Share Option Plan (3) | |
| 4.15 | Can-Fite BioPharma Ltd. 2013 Israeli Share Option Plan (4) | |
| 4.16 | Form of Securities Purchase Agreement dated as of March 10, 2014 between Can-Fite BioPharma Ltd. and the investors listed therein (5) |
| 121 |
| 4.17 | Form of Warrant dated March 10, 2014 issued by Can-Fite BioPharma Ltd. (5) | |
| 4.18 | Form of Registration Rights Agreement dated as of March 10, 20142014 between Can-Fite BioPharma Ltd. and the investors listed therein (5) | |
| 4.19 | Form of Lock-Up Agreement dated March 10, 2014 between Can-Fite BioPharma Ltd. and officers and directors of Can-Fite Biopharma Ltd. (5) | |
| 4.20 | Form of Placement Agent Warrant dated March 10, 2014 issued by Can-Fite BioPharma Ltd. to Roth Capital Partners, LLC(5) | |
| 4.21 | Form of Securities Purchase Agreement dated as of December 2, 2014 between Can-Fite BioPharma Ltd. and the investors listed therein (6) | |
| 4.22 | Form of Warrant issued by Can-Fite BioPharma Ltd. on December 8, 2014 (6) | |
| 4.23 | Letter Agreement between H.C. Wainwright & Co., LLC and Can-Fite BioPharma Ltd. dated December 2, 2014 (6) | |
| 4.24 | Distribution and Supply Agreement between Can-Fite BioPharma Ltd. and Cipher Pharmaceuticals Inc. dated as of March 20, 2015 (4)† | |
| 4.25 | Form of Securities Purchase Agreement dated as of September 19, 2015 between Can-Fite BioPharma Ltd. and the investors listed therein (7) | |
| 4.26 | Form of Warrant issued by Can-Fite BioPharma Ltd. on September 21, 2015 (7) | |
| 4.27 | Letter Agreement between H.C. Wainwright & Co., LLC and Can-Fite BioPharma Ltd. dated September 18, 2015 (7) | |
| 4.28 | Form of Securities Purchase Agreement dated as of October 13, 2015 between Can-Fite BioPharma Ltd. and the investors listed therein (8) | |
| 4.29 | Form of Warrant issued by Can-Fite BioPharma Ltd. on October 15, 2015 (8) | |
| 4.30 | Letter Agreement between H.C. Wainwright & Co., LLC and Can-Fite BioPharma Ltd. dated October 13, 2015 (8) | |
| 4.31 | Distribution Agreement between Can-Fite BioPharma Ltd. and Chong Kun Dang Pharmaceutical Corp. dated as of October 25, 2016 *† | |
| 4.32 | Form of Securities Purchase Agreement dated as of January 18, 2017 between Can-Fite BioPharma Ltd. and the investors listed therein (9) | |
| 4.33 | Form of Warrant issued by Can-Fite BioPharma Ltd. on January 18, 2017 (9) | |
| 4.34 | Letter Agreement between H.C. Wainwright & Co., LLC and Can-Fite BioPharma Ltd. dated January 18, 2017 (9) | |
| 8.1 | List of Subsidiaries of Can-Fite BioPharma Ltd. * | |
| 12.1 | Certification by Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002* | |
| 122 |
| 12.2 | Certification by Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002* | |
| 13.1 | Certification by Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002* | |
| 13.2 | Certification by Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002* | |
| 15.1 | Consent of Independent Registered Public Accounting Firm.* |
* † |
File Herewith. Certain confidential information contained in this exhibit was omitted by means of redacting a portion of the text and replacing it with [...]. This exhibit has been filed separately with the Commission without the redaction pursuant to a Confidential Treatment Request under Rule 24b-2 of the Securities Exchange Act of 1934 and Rule 406 of the Securities Act. |
| (1) | Incorporated herein by reference to Form F-3 filed with the SEC on January 19, 2016. |
| (2) | Incorporated herein by reference to the Registration Statement on Form 8-A filed with the SEC on November 15, 2013. |
| (3) | Incorporated herein by reference to Amendment No. 1 to the Draft Registration Statement on Form 20-F filed with the SEC on September 10, 2013. |
| (4) | Incorporated herein by reference to Annual Report on Form 20-F filed with the SEC on March 27, 2015. |
| (5) | Incorporated herein by reference to the Current Report on Form 6-K filed with the SEC on March 10, 2014. |
| (6) | Incorporated herein by reference to the Current Report on Form 6-K filed with the SEC on December 4, 2014. |
| (7) | Incorporated herein by reference to the Current Report on Form 6-K filed with the SEC on September 22, 2015. |
| (8) | Incorporated herein by reference to the Current Report on Form 6-K filed with the SEC on October 15, 2015. |
| (9) | Incorporated herein by reference to the Current Report on Form 6-K filed with the SEC on January 20, 2017. |
| 123 |
SIGNATURES
The Registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this Annual Report on its behalf.
| CAN-FITE BIOPHARMA LTD. | ||
| Date: March 30, 2017 | By: | /s/ Pnina Fishman, Ph.D. |
| Pnina Fishman, Ph.D. | ||
| Chief Executive Officer | ||
124